We have seen that most reactions of aromatic compounds involve electrophilic substitutions because the π electrons make the aromatic ring electron-rich and therefore, nucleophilic. However, some aryl halides with a strong electron-withdrawing substituent(s) on the ring can undergo nucleophilic substitution (SNAr) instead of electrophilic substitution:

X here is the leaving group and the EWG stands for an electron-withdrawing group (most often nitro) which is there to activate the ring by making it electron-deficient.
There are two main mechanisms: addition-elimination or elimination-addition, also known as the benzyne mechanism which is sometimes considered as a separate reaction. The benzyne mechanism works mainly with a very strong base and nucleophile, NH2– and does not require an EWG group, although its presence is not a hurdle. Let’s put some common examples of these mechanisms and look into the details of the main patterns.

Nucleophilic Aromatic Substitution – The Addition-Elimination Mechanism
These are the ones in the left column. Notice that the nitro group is typically used as an electron-withdrawing group even though other groups such as carbonyls can also activate the ring toward a nucleophilic attack.
As for the nucleophile, a variety of charged and neutral strong nucleophiles such as –OH, –OR, –SR, NH3, and other amines can be used.
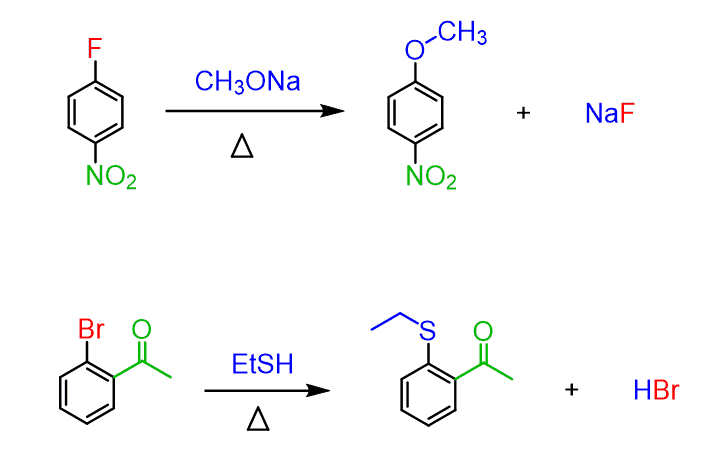
One requirement for these reactions is the ortho or para position of the electron-withdrawing group to the leaving group. This is the only orientation supporting the resonance stabilization of the negative charge in the transition state.
We are going to talk about the details of the mechanism below, but for now, let’s also mention that the reactivities of aryl halides increase, depending on the leaving group, in the following order:

So, the more electronegative the halogen, the better leaving group it is in a nucleophilic aromatic substitution.
And this is the opposite of what we learned in the SN1 and SN2 reactions. We learn that the carbon-fluorine bond is the strongest and the iodide being polarizable was the best leaving group.
What is the reason for this change of reactivity and, in general, what is the mechanism of nucleophilic aromatic substitution?
Let’s now try to answer these important questions.
The Mechanism of Nucleophilic Aromatic Substitution
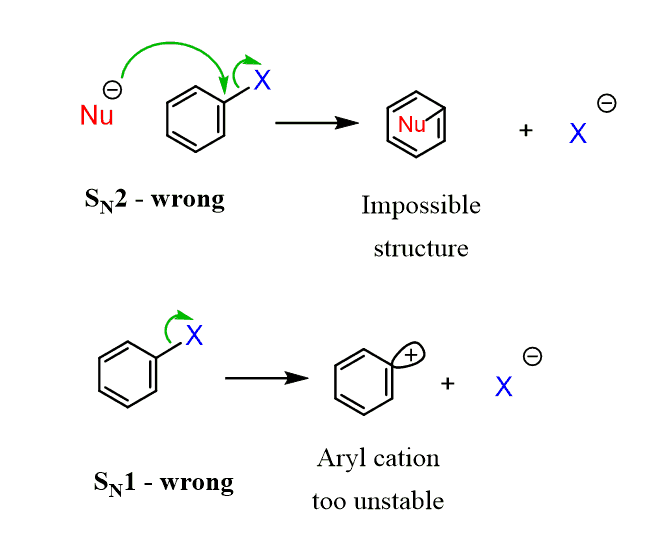
Like in other substitution reactions, the leaving group halide is replaced by a nucleophile. The mechanism of nucleophilic aromatic substitution, however, is different than what we learned in the SN1 and SN2 reactions. The key difference is that aryl halides cannot undergo an SN2 by a backside attack of the nucleophile and, unlike SN1, the loss of the leaving group cannot occur since the phenyl cations are very unstable:
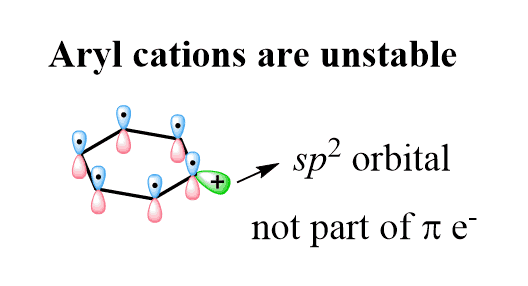
Even though the cation is surrounded by double bonds, it cannot be stabilized because the p orbital, being part of the aromatic ring, is full and the empty orbital is an sp2 orbital perpendicular to the conjugated p orbitals:
The only exception to this is when we have the excellent leaving group of gaseous nitrogen:
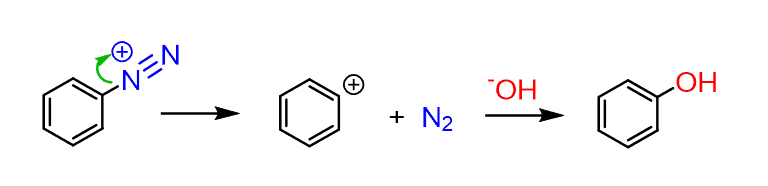
These are the reactions of arene diazonium salts which are sort of unique and represent a good set of strategies in the chemistry of aromatic compounds.
So, no SN1 or SN2 in nucleophilic aromatic substitutions!
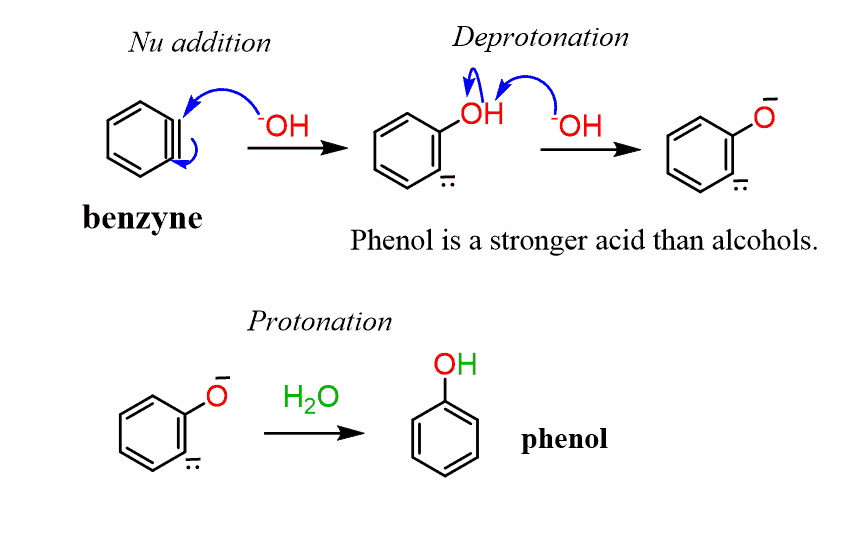
Instead, the reaction occurs either by an addition-elimination mechanism. It starts with the addition of the nucleophile to the aromatic ring generating a resonance-stabilized carbanion.
This addition occurs from either face, almost perpendicular, of the aromatic ring because that’s where the LUMO orbitals are. The resonance-stabilized carbanion is called a Meisenheimer complex:
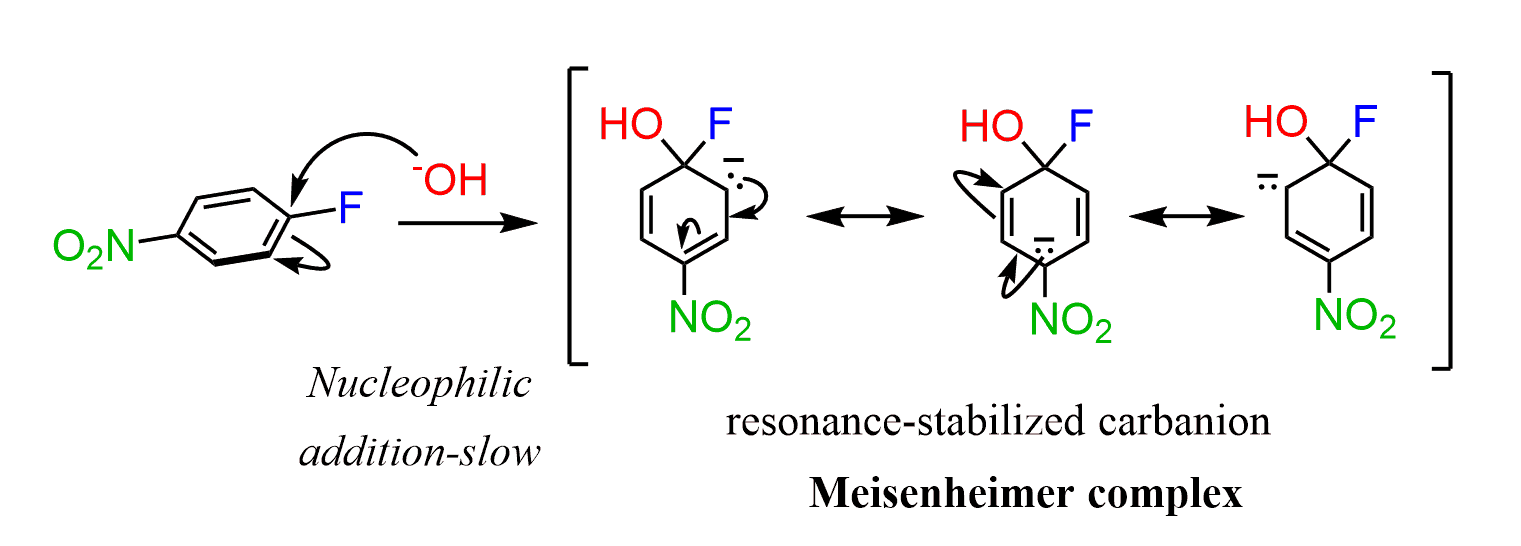
In the next step, the leaving group is eliminated, restoring the aromaticity of the ring:
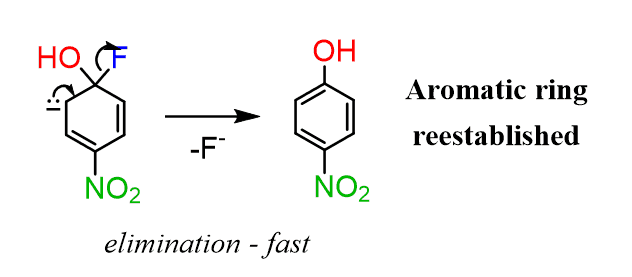
You may wonder why the ring did not kick out the nucleophile instead of the halide. And this simply has to do with their stability – better leaving group is better stabilized. That is why the nucleophile must be a stronger base than the leaving group in order to favor the desired replacement.
Now, let’s go back to the two important observations and requirements in these reactions;
1) Why must the electron-withdrawing group be in the ortho/para position?
To answer this question, draw the mechanism of nucleophilic aromatic substitution for ortho, meta, and para-fluoronitrobenzene:
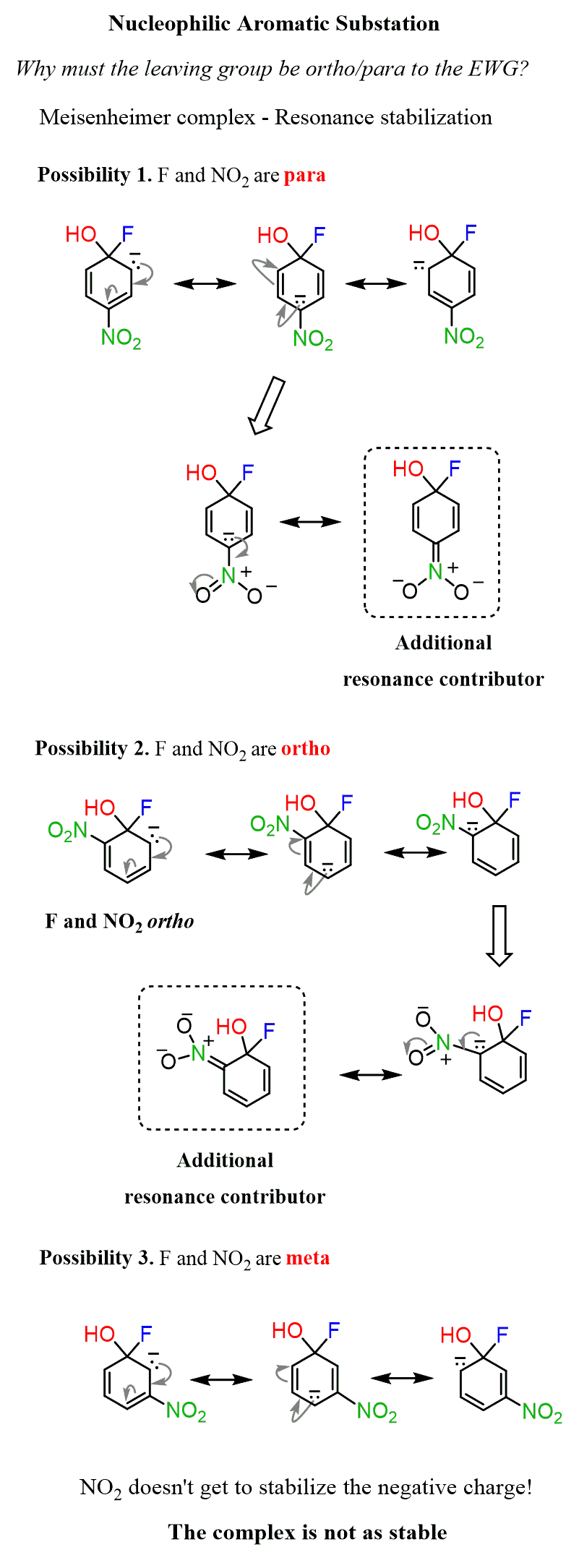
You can see that the electronegative nitro group gets to handle the negative charge by resonance-delocalization only if it is in the ortho or para position. The Meisenheimer complex is not stabilized when the EWG group is meta to the leaving group.
2) Why is the reaction with fluorine the fastest?
The origin of this question is the comparison of nucleophilic aromatic substitution to SN1 and SN2 reactions where the breaking of the C-F bond occurs in the rate-determining step (SN2 is usually one step).
And since the C-F bond is stronger than the other C-halogen bonds, fluoride is the worst leaving group slowing down the substitution.
Now, the difference here is that in the addition-elimination mechanism, the rate-determining step is the nucleophilic addition to the aromatic ring because breaking the aromaticity is energetically very unfavorable:

Therefore, the loss of the fluoride does not significantly slow down the reaction. It still does, compared to if it was an iodide, however, this happens only after the Meisenheimer complex has formed. And again, because the formation of the Meisenheimer complex is the rate-determining step, the nature of the halogen and the C-X bond is not as important as it was in the traditional nucleophilic substitution reactions.
Now, if it is not, then why, for example, iodide is not still better than fluoride, right?
Well, the thing is, we need to also consider how halogens affect the rate of nucleophilic addition in the first step and not just the loss of the leaving group step.
The advantage of fluorine is that being the most electronegative, it activates the ring a lot more than the other halogens by decreasing the electron density and stabilizing the forming negative charge. Again, this is the rate-determining step and it turns out to compensate and overcome the barrier of breaking the C-F bond.
The Benzyne Mechanism in Nucleophilic Aromatic Substitution
Let’s now discuss the second mechanism by which nucleophilic aromatic substitutions occur: the elimination-addition mechanism.
The reaction with –OH requires extreme conditions, however, works at lower temperatures with NaNH2 because it is a very strong base and the reaction goes through an elimination forming a triple-bonded benzyne intermediate.

First, we have a two-step β elimination. The β is the ortho position for aromatic compounds and that’s the most acidic proton which is eliminated by –NH2 or other bases in the first step. The negative charge then is stabilized by the inductive effect of the halogen which is eventually kicked out by this lone pair generating the highly reactive benzyne intermediate.
The rate-determining step of the reaction depends on the halogen. According to March’s Advanced Chemistry, when the leaving group is Br or I, proton removal is rate-determining and the rate order for this step is F>Cl>Br>I. When Cl or F is the leaving group, breaking the C-X bond, or the formation of the benzyne intermediate, is rate-determining, and
the order for this step is I>Br>Cl>F. Notice that regardless of the halogen, deprotonation of the aromatic ring as the resulting carbanion is stabilized by the inductive effect of the halogen rather than a resonance stabilization.
Benzyne is so reactive that it reacts with any available nucleophile to release the strain of a triple bond in the ring. The resulting anion is then protonated by almost any proton source.
Now, compared to this, the transformation with hydroxide is a lot more difficult to accomplish:
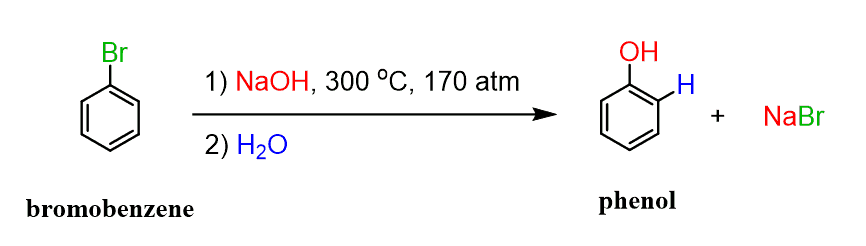
Notice from the list of reactions above that when there is an electron-withdrawing group on the aromatic ring, the conditions are incomparably milder. The mechanism is the same as with sodium amide.
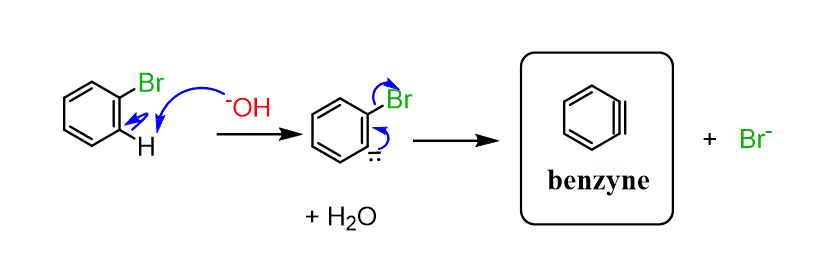
So, one important detail in the mechanism that we must discuss is: how is that triple bond crammed in the ring?
Well, from what we know about the geometries and hybridizations, it just looks bothering to have a triple bond in a six-membered ring where the angles are 120o.
If these two carbons were sp-hybridized as judged from the triple bond and steric number, it would’ve probably been impossible since the sp orbitals are pointing in two linear directions.
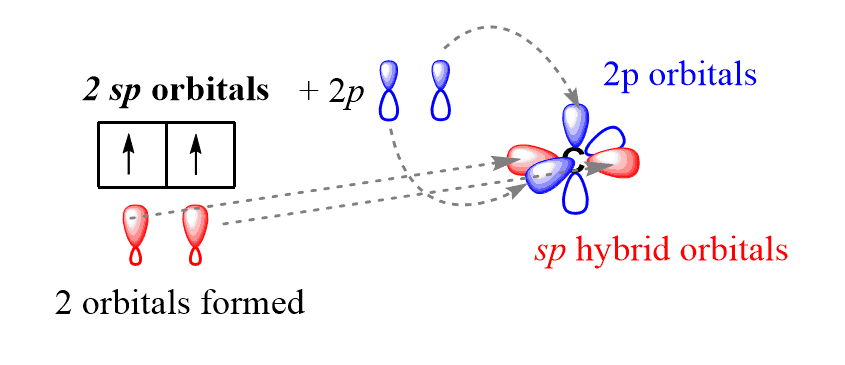
So, this strange triple bond is likely formed between two sp2 hybridized carbons:
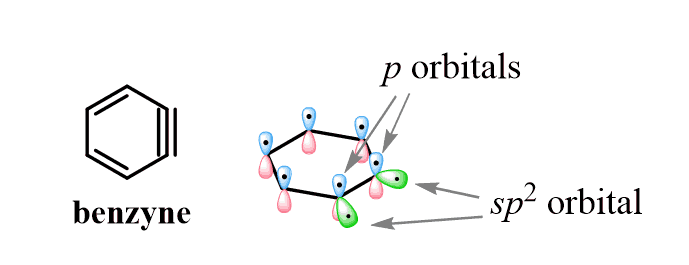
- One of the sp2 orbitals of each carbon makes the single bond
- One of the π bonds is formed by the overlap of the p orbitals, just as it usually is
- And the second π bond is formed by the overlap of two sp2 orbitals
And now a good question about the mechanism itself:
How do we prove the formation of the benzyne intermediate?
One of the methods for proving the formation of benzyne intermediate is using 14C isotope labeling:
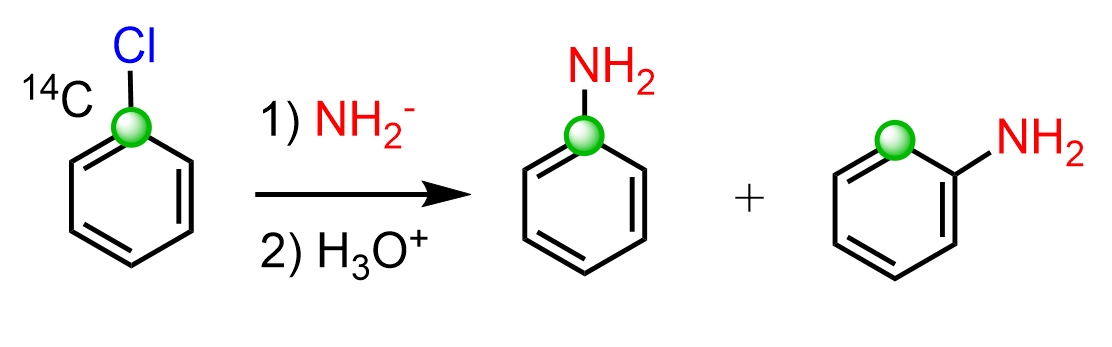
The NH2 wouldn’t end up being on both carbons if there was no benzyne forming. At least, it is a strong suggestion.
The idea can be applied by using a substituted aryl halide. For example, let’s consider the reaction of p-chlorotoluene with NaNH2 that produces a mixture of two constitutional isomers:
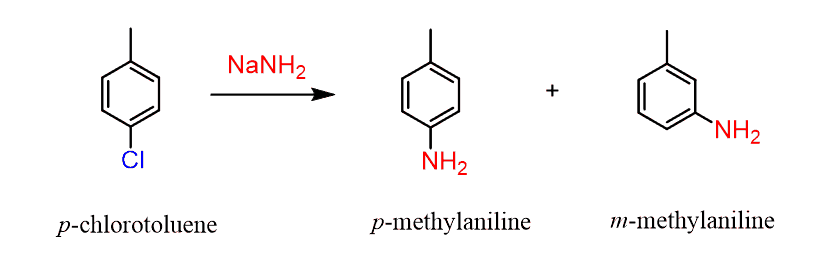
This observation can be explained by the nucleophilic attack of the –NH2 at both carbons of the triple bond of the benzyne intermediate:
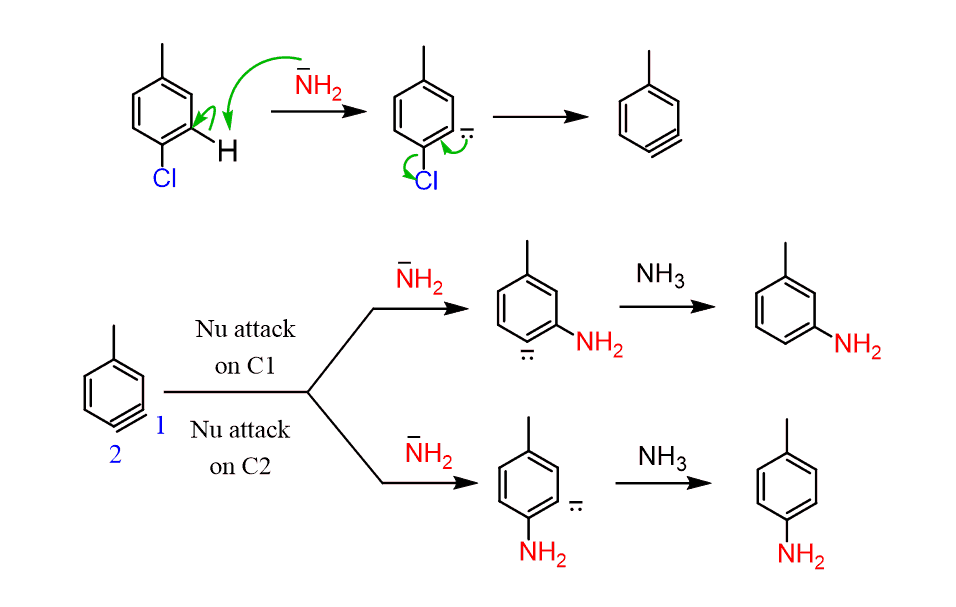
Another, perhaps a more convincing example to prove the formation of benzyne is the reaction of sodium amide with a strong electron-withdrawing group next to the halogen:
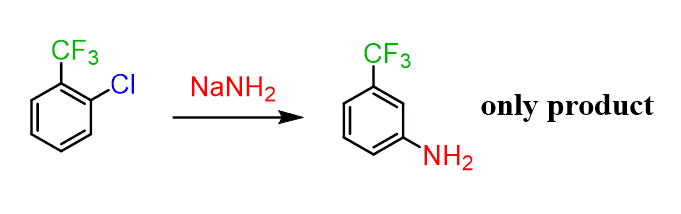
What is important here is the regiochemistry of the reaction. Unlike the previous example, there is only one product formed in this reaction.
And this can be explained by the selective addition of the amide nucleophile to the benzyne such that only the more stable carbanion is formed:

The first carbanion is stabilized by the highly electronegative trifluoromethyl group through an inductive effect since the electron pair in the sp2 orbital does not overlap with the π orbitals of the aromatic system and mesomeric effects are not relevant here.
And one more, beautiful, evidence of the benzyne-forming mechanism is the trapping experiment.
We add furan to the reaction mixture, and it acts as a diene trapping the benzyne intermediate in a Diels-Alder reaction:
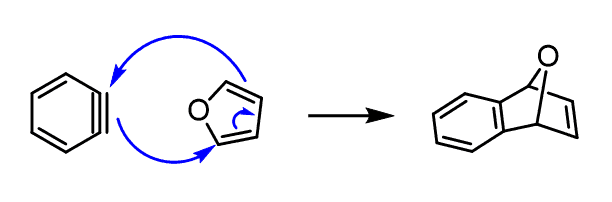
The Regiochemistry of Nucleophilic Aromatic Substitution
For the traditional SNAr, it is easy as the nucleophile appears where the leaving group was.
For the benzyne mechanism, remember this: When no electron-withdrawing group is present on the ring, a mixture of isomers is obtained, except if there are no ortho hydrogens next to the halogen (no acidic proton) in which case the reaction does not occur. If the ring contains an inductively EWG, the nucleophile ends up on the carbon that is farther away from the EWG group.

To make a shortcut for predicting the regiochemistry of nucleophilic aromatic substitution via the benzyne intermediate, remember that the nucleophile ends up on the carbon which is farther away from the EWG group. The cause of this selectivity is the tendency of the EWG to hold the negative charge closer which stabilizes the intermediate.
For the ortho and para orientation of the halide and the EWG group, the triple bond can only be formed in one position, so we don’t need to worry about that part. And once the benzyne is formed, the nucleophile attacks the carbon that puts the negative charge closer to the EWG (see the blue colors of the EWG and X in the diagram).
It is important to mention that only inductively EWGs are relevant here as the electrons of the anion are at 90o to the ring and resonance effects cannot be communicated through the aromatic system. Therefore, OCH3, for example, is electron-withdrawing equivalent to CF3!
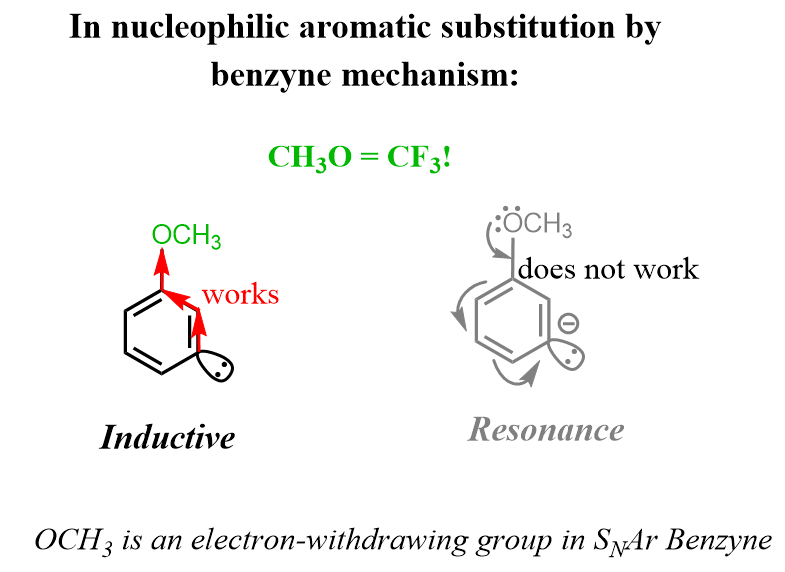
For the meta orientation of the EWG and LG, the base deprotonates the ortho hydrogen as it is the closest to the EWG and the most acidic. This is the rate-determining step and therefore, dictates the regiochemistry of the reaction. Once the triple bond is formed, the nucleophile tends to attack the meta position because a) it puts the negative charge closer to the EWG, b) the ortho position is sterically hindered by the EWG:
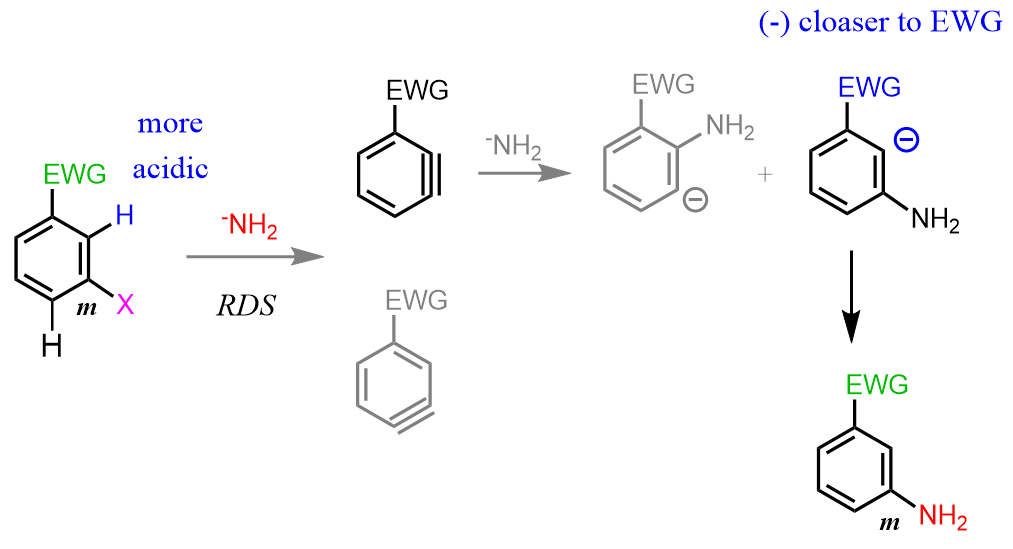
Electron-donating groups do not enforce regioselectivity as much, however, opposite trends will be expected.
This post has been a long one and I wanted to mention great resources such as the book Modern Physical Organic Chemistry by Eric V. Anslyn, March’s Advanced Organic, and Master Organic Chemistry.
Here are some more examples to work on and the answers can be found under the Nucleophilic Aromatic Substitution Practice Problems post.
Practice
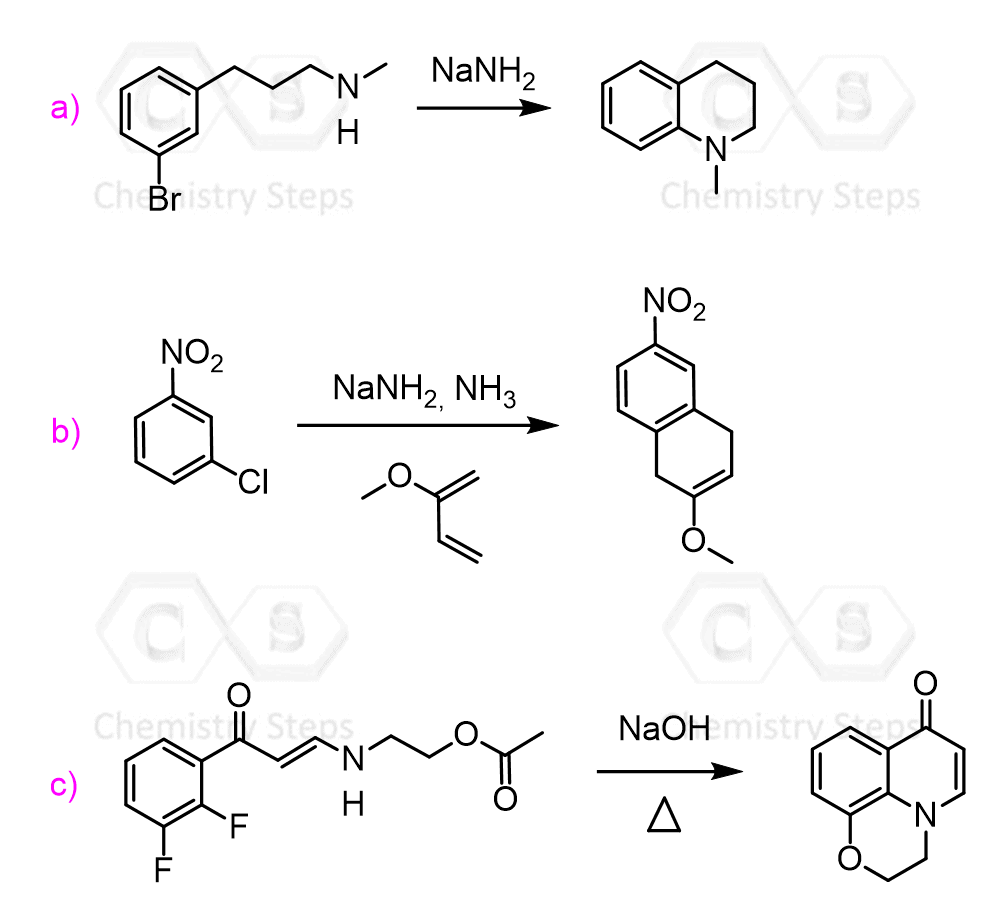
Determine the product in each nucleophilic aromatic substitution reaction.

Draw a possible mechanism for each synthetic transformation:
Check Also
- Naming Aromatic Compounds
- Introduction to Aromatic Compounds
- Benzene – Aromatic Structure and Stability
Aromaticity and Huckel’s Rule - Identify Aromatic, Antiaromatic, or Nonaromatic Compounds
- Frost Circle
- Annulenes
- Electrophilic Aromatic Substitution – The Mechanism
- The Halogenation of Benzene
- The Nitration of Benzene
- The Sulfonation of Benzene
- Friedel-Crafts Alkylation with Practice Problems
- Friedel-Crafts Acylation with Practice Problems
- Vilsmeier-Haack Reaction
- The Alkylation of Benzene by Acylation-Reduction
- Ortho Para Meta in EAS with Practice Problems
- Ortho Para and Meta in Disubstituted Benzenes
- Why Are Halogens Ortho-, Para- Directors yet Deactivators ?
- Limitations of Electrophilic Aromatic Substitution Reactions
- Orientation in Benzene Rings With More Than One Substituent
- Synthesis of Aromatic Compounds From Benzene
- Arenediazonium Salts in Electrophilic Aromatic Substitution
- Reactions at the Benzylic Position
- Benzylic Bromination
- Nucleophilic Aromatic Substitution Practice Problems
- Reactions of Phenols
- Reactions of Aniline
- Meta Substitution on Activated Aromatic Ring
- Electrophilic Aromatic Substitution Practice Problems
- Aromatic Compounds Quiz
Hi 🙂
Thanks for an amazing explanation!
However, I think the correct answers for the practise exercises are gone.. It is possible for you to upload these?
Thanks for an amazing page. If I pass my organic chemistry course, it will be thanks to you!!
Hello,
The answers can be found under the Nucleophilic Aromatic Substitution Practice Problems post.
I hope everything works out on the test.
Good luck!