 Mohamed Amer Musrati
Mohamed Amer Musrati Patrick De Baetselier
Patrick De Baetselier Kiavash Movahedi
Kiavash Movahedi Jo A. Van Ginderachter
Jo A. Van Ginderachter- 1Lab of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Brussels, Belgium
- 2Myeloid Cell Immunology Lab, VIB Center for Inflammation Research, Brussels, Belgium
- 3Lab of Molecular and Cellular Therapy, Vrije Universiteit Brussel, Brussels, Belgium
The liver is a vital metabolic organ that also performs important immune-regulatory functions. In the context of infections, the liver represents a target site for various pathogens, while also having an outstanding capacity to filter the blood from pathogens and to contain infections. Pathogen scavenging by the liver is primarily performed by its large and heterogeneous macrophage population. The major liver-resident macrophage population is located within the hepatic microcirculation and is known as Kupffer cells (KCs). Although other minor macrophages reside in the liver as well, KCs remain the best characterized and are the best well-known hepatic macrophage population to be functionally involved in the clearance of infections. The response of KCs to pathogenic insults often governs the overall severity and outcome of infections on the host. Moreover, infections also impart long-lasting, and rarely studied changes to the KC pool. In this review, we discuss current knowledge on the biology and the various roles of liver macrophages during infections. In addition, we reflect on the potential of infection history to imprint long-lasting effects on macrophages, in particular liver macrophages.
Liver macrophage heterogeneity and functions during homeostasis
Ontogeny of Kupffer cells
The liver harbors a large and heterogeneous population of resident macrophages (Figure 1). Work on mouse models has generated extensive information on the major resident macrophage population in the liver, known as KCs, which resides in the sinusoidal vessels and reaches into the perisinusoidal space of Disse towards hepatic stellate cells and hepatocytes (2). KCs are established embryonically and originate from yolk sac erythro-myeloid progenitors that seed the fetal liver by E10.5, later becoming heavily diluted by fetal liver monocytes (3–6), while also receiving a minor contribution from blood monocytes during the post-natal period (7). Studies have highlighted the importance of the transcription factors Id3 and Nr1h3 (coding for LXRα) for normal KC development and maintenance of identity, respectively (8, 9).
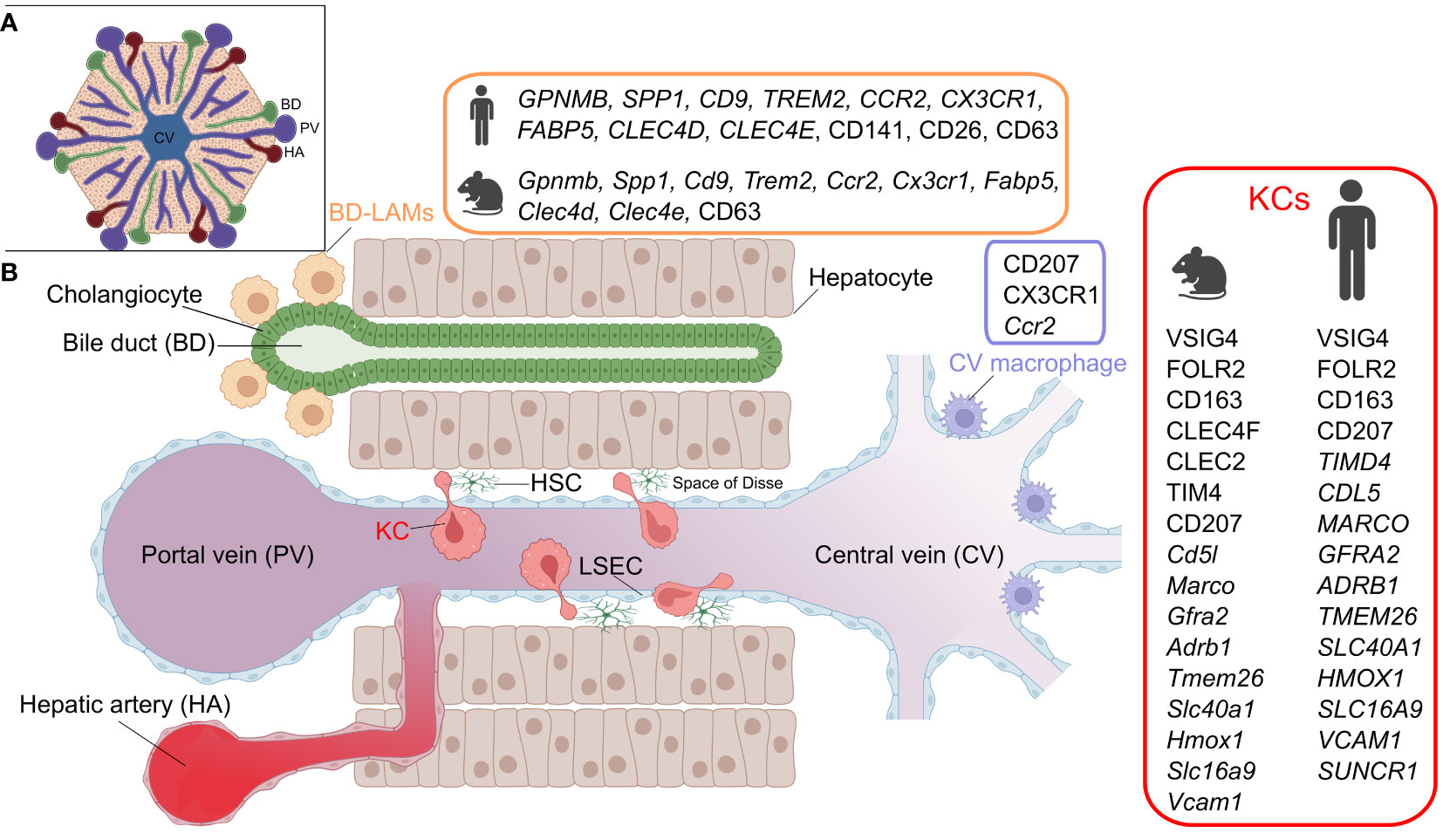
Figure 1 Hepatic macrophage subsets. A) The structure of the liver lobule. B) A cross section of liver sinusoids illustrating the different resident-macrophage populations. KC, Kupffer cell; LAM, lipid-associated macrophage; LSEC, liver sinusoidal endothelial cell; HSC, hepatic stellate cell. Marker details adapted from (1). Figure created with BioRender.com.
During homeostatic conditions, KCs self-maintain and are only marginally replenished by adult bone marrow-derived monocytes (3, 5, 10). However, as will be discussed in later sections, bone marrow contribution to KCs increases dramatically in response to certain liver insults that trigger KC depletion. An important question is whether ontogeny is important for KC functionality. Work utilizing a murine model of diphtheria toxin-mediated specific KC depletion (Clec4f-DTR mice), demonstrated that circulating monocytes engraft in the liver upon KC depletion and give rise to monocyte-derived KCs (moKCs) that are highly reminiscent of their embryonic KC (emKCs) predecessors, at least at the transcriptome level (7). Monocyte engraftment as moKCs is orchestrated by the cellular niche of KCs, comprised of hepatocytes, liver sinusoidal endothelial cells (LSECs), and hepatic stellate cells and mediated via niche-derived signals such as DLL4, bone morphogenetic proteins (BMPs), TGFβ and desmosterol (2, 11). In particular, the ALK1-BMP9/10 axis, involving ALK1 (KCs) and BMP9/10 (stellate cells), is of critical importance for KC homeostasis, as ALK1 loss leads to defects in KC development and maintenance (12, 13).
Phenotypically, murine KCs can be readily identified by their unique surface expression of the endocytic C-type lectin receptor CLEC4F (7, 14). Additionally, KCs can be distinguished from other liver macrophages by TIM4, CLEC1B and VSIG4 expression, although these markers can also be expressed by other tissue-resident macrophages outside the liver (14, 15). One outstanding current challenge is to phenotypically distinguish between moKCs and emKCs. Given that TIM4 is acquired in the late stages of KC development, several studies relied on the lack of TIM4 expression to define moKCs in models of non-alcoholic steatohepatitis and irradiation-induced liver injury (7, 16–18). However, it is likely that the temporal regulation of TIM4 acquisition is model-dependent. Indeed, in the context of natural KC development during embryogenesis, Timd4 expression occurs relatively early and precedes the upregulation of other KC marker genes such as Clec4f (2). Likewise, Cd163 mRNA has been shown to be mainly enriched in emKCs (7, 17). However, in situ translatome analysis using a Cre-driven RiboTag reporter revealed that emKCs and moKCs express similar levels of CD163 (19). One marker that appears to be highly enriched in emKCs, and lowly expressed by moKCs across models is MARCO (17, 19). However, whether MARCO expression by itself enables a clear distinction between the two subsets has not been tested. Consequently, genetic fate-mapping, using transgenic mouse strains that report bone marrow ontogeny, remains the most reliable and non-invasive method for separating em- and moKCs.
Functions of Kupffer cells
KCs perform a wide array of homeostatic functions that support liver functionality and limit tissue injury. KCs play a key role in maintaining immunological tolerance in the liver under baseline conditions. For instance, human KCs secrete IL-10 in response to LPS stimulation in vitro (20). Murine KCs scavenge circulating particles, promote the expansion of Foxp3+IL-10+ T regulatory cells (21) and suppress dendritic cell-induced T-cell activation by the action of prostaglandins (22). Moreover, the liver is a major site for iron metabolism (23) and murine KCs are implicated in regulating iron homeostasis as they express key genes involved in macrophage heme-iron metabolism, such as Spi-c and Nfe2l2 (24). Murine KCs scavenge damaged and aged red blood cells and are strategically located to deliver iron to hepatocytes (2, 24, 25). Murine KCs are also thought to control body iron levels by regulating hepcidin expression (26), although this remains controversial (27). In addition to their erythro-phagocytic capacity, murine KCs are implicated in maintaining hemostasis by contributing to the clearance of aged platelets (28, 29). This is mediated by the Ashwell-Morell receptor and macrophage galactose lectin, as well as CLEC4F (28, 29). As such, KC depletion in mice via clodronate liposomes was associated with adverse platelet dysfunctions, as manifested by increased bleeding following wounding (29). Furthermore, murine KCs participate in the efferocytosis of apoptotic cells (30), and are also thought to regulate circulating neutrophil numbers by mediating the clearance of apoptotic neutrophils (31, 32). KCs are also implicated in lipid metabolism as they are enriched in the expression of liver X-family of receptors (LXRs) and peroxisome proliferator-activated family of receptors (PPARs), involved in various aspects of cholesterol transport and metabolism (reviewed in (33)). Murine KCs are abundantly equipped with low density lipoprotein (LDL) receptor, and appear to serve as gatekeepers for LDL cholesterol transport to hepatocytes (34). In line with this, in Csf1r knockout rats, which exhibit major deficiencies in many tissue-resident macrophages including KCs, the liver shows a vast dysregulation of genes related to lipid metabolism, liver growth and function, and endures extensive age-progressive steatosis (35). Additionally, murine KCs have recently been shown to be the major source of miR-690 within the liver, a microRNA that negatively regulates hepatocyte lipogenesis and hepatic stellate cell fibrogenic activation (36). Deletion of miR-690 shifts KCs towards a pro-inflammatory polarization, suggesting that this microRNA regulates some KC functions in the steady state liver (36). Consistent with this, treatment with miR-690 can reverse the impaired erythro-phagocytic capacity of KCs associated with non-alcoholic steatohepatitis (36).
Liver macrophage heterogeneity
A recent study has identified two distinct subpopulations of embryonically-derived KCs in mice: a major CD206lowESAM– population (KC1) and a minor CD206highESAM+ population (KC2) (37). Although they share the expression of core KC genes, KC1 mainly expresses genes associated with immune functions, while KC2 exhibits a higher expression of endothelial markers and genes associated with metabolic pathways such as lipid metabolism. Accordingly, the depletion of KC2 has been shown to prevent diet-induced obesity (37). Subsequent reports argued that the KC2 might represent endothelial cell-KC doublets, hence, these populations are still a current subject of debate (12, 38, 39).
Although KCs represent the major population of liver-resident macrophages, the steady state liver also hosts other macrophage populations that are spatially segregated (Figure 1) (12). These include bile duct-associated macrophages, which show transcriptional similarities to lipid-associated macrophages, expressing genes such as Gpnmb, Spp1 and Trem2 (12). Notably, a similar counterpart is also present in human livers (12). Moreover, another population of Cd207-expressing macrophages has been detected around the central veins of the murine liver (12). Apart from some data suggesting that murine bile duct macrophages exhibit a lower pro-inflammatory response to LPS stimulation than KCs, these newly identified macrophages generally lack functional characterization (12). Finally, the murine liver also harbors another population of macrophages residing in its capsule (40). These CX3CR1-expressing capsular macrophages are derived from monocytes, and accumulate after weaning in a microbiota-dependent manner and perform defensive functions against peritoneal pathogens (40).
Human Kupffer cells
A number of studies sought to characterize KCs in human livers (1) and we hereby review notable findings. Single cell RNA-sequencing (scRNA-seq) of CD163+VSIG4+ cells from human livers has revealed subpopulations with distinct gene expression signatures, including MARCO+LILRB5+ and CD1C+FCER1A+ populations, enriched in metabolic/immunoregulatory and antigen presentation pathways, respectively (41). Another study also reported a VSIG4+MARCO+ population based on scRNA-seq of human livers and has histologically shown that these cells are concentrated in periportal areas (42). Moreover, analysis of healthy and cirrhotic human livers identified two KC-like populations that were commonly enriched in CD163, MARCO and CD5L, while differing in TIMD4 expression. Noteworthy, the MARCO+TIMD4- population was less present in the cirrhotic livers. Immunohistochemistry showed that while TIMD4+ cells were equally present in both the healthy and cirrhotic livers, MARCO+ cells were reduced during cirrhosis (43). A recent analysis of healthy human livers, using spatial proteo-genomics and single-cell sequencing approaches, identified a macrophage population that resembles murine KCs (12). The human KC-like population highly expressed core murine KC markers, including CD5L, VSIG4, SLC1A3, CD163, FOLR2, TIMD4, MARCO, GFRA2, ADRB1, TMEM26, SLC4OA1, HMOX1, SLC16A9 and VCAM1 (Figure 1), and appears to be localized in the mid zones of the liver lobules. Additionally, the same study detected a moKC-like hepatic macrophage subset in human livers, which expresses many of the core KC genes but lacks TIMD4 expression (12). It is noteworthy that human KCs lack the expression of the specific murine KC marker CLEC4F, and in contrast to murine counterparts, express SUCNR1 (12) (discrepancies in KC markers between species and studies were recently reviewed in (1)). Although delineating the ontogeny of KCs in humans is difficult to achieve, it is clear that humans generally endure a variety of pathological insults that affect the liver throughout life. Several insults that are known to alter KC ontogeny in mice, such as infections, a high-fat diet and alcohol consumption, are highly relevant for humans. It is therefore likely that the ontogeny of human KCs varies across individuals, and may possibly be a mixture of bone marrow-derived as well as embryonic KCs. Variability likely also exists at the level of the activation status of human KCs since exposure to inflammatory episodes can imprint prolonged changes on macrophages, as elaborated later in this review.
Liver macrophages in the context of infections
Bacterial infections
The liver has an outstanding capacity to filter circulating bacteria (44, 45). This superior microbial scavenging property is underlined by the presence of KCs in the hepatic vasculature (44, 46). Due to their remarkable pattern of zonation around the periportal tracts of the liver blood vessels (47), KCs are among the first cells to encounter the incoming antigen-rich portal blood. KCs form an extensive scavenger network that actively monitors sinusoidal blood and captures circulating bacteria (48–50). KCs are equipped with a large repertoire of phagocytic receptors that facilitates the clearance of bacterial pathogens (49–51). A well characterized KC receptor that plays an essential role in bacterial clearance is VSIG4 (48–50). In addition to serving as a complement receptor for C3-opsonized bacteria (50), VSIG4 can directly bind the lipoteichoic acid component of the gram-positive bacterial cell wall, enabling their phagocytosis by KCs (48). Indeed, loss of Vsig4 results in enhanced bacterial dissemination and increased mortality in mice infected with Listeria monocytogenes (50). Deletion of Vsig4 in a mouse model of alcoholic liver disease hinders the phagocytosis of Enterococcus faecalis by KCs, and associates with increased systemic dissemination of gut-translocated bacteria as well as exacerbated pathology (52). Noteworthy, decreased Vsig4 expression and a reduced percentage of VSIG4+ macrophages is seen in the livers of patients with alcohol-related liver disease (52). Aside from the direct role in bacterial clearance via phagocytosis, KCs can indirectly instruct other innate immune cells to launch anti-microbial responses. For example, during Borrelia burgdorferi infection, murine KCs present antigen to invariant natural killer T cells (iNKT) in a CD1d-dependent manner, leading to iNKT cell activation and interferon-γ production (46). Activated iNKT cells in turn restrict the extra-hepatic dissemination of bacteria (46). Furthermore, the capture of certain bacterial pathogens such as Bacillus cereus and Methicillin-resistant Staphylococcus aureus (MRSA) by murine KCs has been shown to induce platelet aggregation on KCs (53). This process requires interactions between GPIb and GPIIb on platelets with von Willebrand factor (vWF) on KCs. Platelet aggregation appears to serve an important protective function in response to B. cereus infection in mice as GPIbα deficiency leads to increased mortality, impaired bacterial clearance, exacerbated liver damage and vascular injury (53). Interestingly, murine KCs possess dual-track mechanisms to remove bacteria that permit either the slow or fast clearance of bacterial pathogens (49). The former clearance route is regulated by time-dependent opsonization of bacteria by platelets via interaction with complement proteins. These bacteria-platelet complexes exhibit a relatively increased half-life in the circulation and are subsequently captured by VSIG4 on KCs. Conversely, the fast clearance route is mediated by scavenger receptors in a platelet-independent manner, hence allowing the rapid removal of bacteria (49). This dual-track clearance mechanism ensures the balance between rapid removal of bacteria while also maintaining antigen availability for the priming of adaptive immune responses (49). Interestingly, the anti-bacterial properties of murine KCs are strongly regulated by the gut microbiome (particularly the bacterial microbiome), and depletion of the gut microbiota associates with defective KC responses and results in higher vulnerability to infection (47, 54). Remarkably, the spatial distribution of KCs around the periportal tracts in the liver is in itself microbiota-dependent (47). Mechanistically, microbiome-derived signals regulate the composition of the extracellular matrix of LSECs in a MyD88-dependent manner, causing CXCL9 retention on periportal LSECs, hence allowing interaction with CXCR3 on KCs (47). Moreover, continuous crosstalk with gut commensals can directly shape the bacterial killing properties of KCs in a microbiome-derived, D-lactate-dependent manner (54). In addition, murine KCs exhibit a loss of their typical phenotype, gene signature and spatial zonation upon the deletion of Alk1, and this associates with impaired bacterial clearance and ultimately increased mortality in response to bacterial infection (13).
Intriguingly, encounter with some bacterial pathogens seems to compromise KC viability, resulting in the death and transient depletion of KCs. For instance, the facultative intracellular bacterial pathogens L. monocytogenes, as well as Salmonella enterica serovar Typhimurium, are known to infect murine liver macrophages leading to macrophage death (55). KCs respond to L. monocytogenes infection by undergoing necroptosis (55), a form of programmed lytic cell death via Receptor-Interacting Protein Kinase 3 (RIPK3) activation of Mixed Lineage Kinase Domain-like (MLKL) (56). Dying KCs become massively replaced by CCR2-dependent, monocyte-derived macrophages that proliferate intrahepatically, driven by signals including CSF1, hepatocyte-derived IL-33 and basophil-derived IL-4 (55). Interestingly, L. monocytogenes-induced KC necroptosis is thought to serve a beneficial host response by orchestrating a type 2 inflammation that limits excessive liver injury. Mechanistically, the necroptosis of KCs promotes hepatocyte production of IL-33, which presumably induces IL-4 production by basophils and thereby the alternative activation of liver macrophages (55). Paradoxically, another study reported that L. monocytogenes induces rapid cell death in murine liver macrophages in an IRF3-dependent manner, and showed that Irf3 deletion rescues KC death and reduces hepatic bacterial burden (57), suggesting a pathogenic consequence associated with KC death in this context. Notably, no restoration of emKCs is observed following L. monocytogenes infection, and instead, recruited moKCs appear to dominate and engraft durably in the liver after the clearance of infection (55). Whether this change alters KC responses to subsequent bacterial infections is unknown. However, murine moKCs have been shown to exhibit a markedly superior phagocytosis of a number of bacterial pathogens, including L. monocytogenes (17), suggesting that the modulation of KC ontogeny might lead to functional consequences. In contrast, in vitro examination has shown that moKCs generated in the Clec4f-DTR mouse model are equally phagocytic as emKCs (7). Nevertheless, the latter finding might reflect functional changes associated with the loss of the natural KC microenvironment, or even the artificial, non-physiological method of moKCs generation. Noteworthy, the phagocytic capacity of KCs can be altered by exposure to environmental factors. For instance, certain clinical drugs have been shown to negatively modulate the scavenging properties of murine KCs (58, 59). For example, tacrolimus, a commonly used drug to prevent graft rejection after solid organ transplantation and to treat graft-versus-host disease after bone marrow transplantation, has been shown to inhibit bacterial uptake and killing by KCs in mice (59). In humans, the risk of Staphylococcus aureus infection is associated with higher tacrolimus concentrations in liver transplant recipients (59). An overdose of acetaminophen has also been shown to imprint an immune-suppressive profile on murine KCs by upregulating PD1 at the cell surface, and to impair their phagocytic properties (58). Intriguingly, contrasting the negative effect of the aforementioned drugs on KCs, physical exercise has been shown to boost the phagocytic capacity of murine KCs, and to increase their expression of the phagocytic receptors MARCO and SR-A, which associates with enhanced endotoxin clearance from the blood circulation (60). Moreover, exercise training imprints a long-lasting anti-inflammatory profile on murine KCs, manifested in a lowered production of pro-inflammatory cytokines and an enhanced secretion of IL-10 and IL-1Ra in response to an LPS challenge (61). This exercise-induced anti-inflammatory reprogramming of KCs is regulated by itaconate metabolism, and can be recapitulated by KC treatment with itaconate, whereas the deletion of Irg1 (which codes for cis-aconitate decarboxylase) abrogates this beneficial effect of exercise in mice (61). Importantly, the livers of exercised mice undergo less injury and necrosis following an LPS challenge (61), thus highlighting the effects of physical exercise in modulating the response to infectious inflammation in the liver. Aside from the various roles played by KCs during microbial infections, murine liver capsular macrophages are also implicated in the response to bacteria. They are thought to limit peritoneal pathogen dissemination to the liver by recruiting neutrophils, although the underlying mechanism remains vague (40).
In summary, in addition to their typical scavenging role, KCs regulate complex processes that facilitate the containment of bacterial infections and tissue injury. Moreover, the bactericidal properties of KCs are subject to regulation by environmental factors.
Viral infections
Hepato-tropic viruses, such as hepatitis B virus (HBV), can establish life-long persistent infections (62) and KCs seem to be implicated in the different aspects of HBV pathogenesis. Mouse experiments utilizing clodronate liposome-mediated KC depletion have suggested that KCs exhibit suppressive effects on CD8+ T cells during HBV infection (63). As such, KC depletion lessens HBV persistence and associates with enhanced numbers and activation of CD8+ T cells (63). Interestingly, KC-mediated suppression of CD8+ T cells has been shown to be TLR2-dependent, as KCs from WT but not TLR2 KO mice abundantly produced the CD8+ T cell-suppressive cytokine IL10 following in vitro stimulation with the hepatitis B core antigen (63). Murine KCs have also been reported to use an IL-10-dependent mechanism to promote systemic specific humoral tolerance to HBV (64). Recent work further explored the cellular and molecular mechanisms responsible for CD8+ T cell dysfunction during HBV infection. To dissect the consequences of hepatocellular-mediated priming versus KC-mediated priming of CD8+ T cells, a recent study performed cell-selective HBV targeting to either hepatocytes (natural target of HBV) or KCs (non-natural targets of HBV) in mice. This approach revealed that hepatocellular priming of virus-specific CD8+ T cells generates dysfunctional CD8+ T cells with impaired antiviral capacity (65). Conversely, KC-mediated priming leads to CD8+ T-cell differentiation into effector cells endowed with potent antiviral functions (65). KC-primed CD8+ T cells exhibit higher expression of activation markers such as Gzma, Gzmb and Ifng and show increased expression of Il2, the critical T-cell growth factor (65, 66). In line with these observations, treatment with recombinant IL-2 (IL-2-c) rescues and restores the antiviral functions of hepatocellularly-primed murine CD8+ T cells (65). Follow-up work has shown that the ability of IL-2-c treatment to rejuvenate dysfunctional CD8+ T cells during HBV infection is mediated by the KC2 subset, that is enriched in the expression of IL-2 signaling components (67). As such, the depletion of KC2 significantly impairs the efficacy of IL-2-c treatment (67).
Viral infections are also known to elicit dynamic compositional changes in the liver macrophage pool (68, 69). For instance, rapid IRF3-dependent KC death is seen in mice following infection with human adenovirus (57). Intriguingly, KC death during these settings appears functionally relevant. Indeed, although KC death was spared in IRF3 knockout mice, these mice exhibited higher hepatic viral loads. These data suggested either a pathogenic role for KCs during adenovirus infection, or an IRF3-mediated macrophage death pathway that serves a protective role during this infection (57). KC death also occurs after intravenous delivery of adenovirus vectors in mice (70). This was shown to be complement C3-dependent and to require VSIG4 expression, given that KC death is rescued in VSIG4- or C3-deficient mice (71). The process of KC death and replenishment by monocytes has also been explored in the context of vaccinia virus (VACV)-induced acute hepatitis in mice (69). During VACV infection, IFNAR triggering in myeloid cells, including KCs, contributes to the early control of the infection (69). In these settings, KCs are depleted and become replenished by infiltrating monocytes that differentiate into moKCs. Interestingly, bone marrow chimera experiments revealed that IFNAR signaling in monocytes delays their differentiation into moKCs (69). Hence, IFNAR-knockout monocytes engrafted more efficiently in the livers of VACV-infected mice than WT monocytes. This appeared to be a result of a direct IFNAR triggering of monocytes in the liver, as both WT and IFNAR knockout moKCs became equally engrafted in the KC population upon cessation of hepatic IFNAR triggering (69).
In conclusion, KCs clearly perform various and occasionally contrasting roles during viral infections.
Parasitic and fungal infections
KCs are implicated in the pathology of several parasitic infections, and their contribution ranges from beneficial to detrimental. For instance, during experimental trypanosome infections, KCs perform an essential role in parasite clearance, as KC depletion by clodronate liposomes results in uncontrolled parasitemia and rapid mortality (72). In fact, VSIG4 is critical for the anti-trypanosomal response of KCs as it contributes to parasite clearance in a complement C3-dependent manner (72). Furthermore, murine hepatic macrophages, including KCs, are thought to be a source of CXCL16 during Trypanosoma brucei infection, and are hence suspected to be implicated in the recruitment of pathogenic CXCR6+ CD4+ T cells that promote aggravated liver damage and increased mortality during infection (73). Noteworthy, VSIG4-complement interactions seem of crucial importance to universal KC defense responses, since KCs utilize a similar mechanism to contain fungal infections (74). This is highlighted by impaired blood clearance and heightened infection burden in clodronate liposome-treated, as well as VSIG4 knockout or C3 knockout mice following Cryptococcus neoformans infection (74).
Remarkably, some parasites manage to circumvent the clearance by KCs. In case of Plasmodium infection, sporozoites rapidly target the liver to establish hepatocyte infection (75). Strikingly, experimental work has shown that sporozoites traverse through KCs to cross the endothelial barrier and infect hepatocytes (76–78). It has been illustrated that sporozoite crossing of KCs induces extensive KC death, with only a small fraction of KCs appearing to survive the crossing event (78). Although sporozoites can also traverse through LSECs, this process appears less common than KC traversal (78). Consistent with this, in vitro work showed that LSECs are less permissive to sporozoite traversal relative to KCs (76), overall suggesting that sporozoites utilize and favor KC crossing to overcome the sinusoidal lining and establish hepatocyte infection. Furthermore, it appears that sporozoites modulate KC functionalities to favor their intracellular survival, as illustrated by in vitro work showing that sporozoites can suppress the respiratory burst in KCs by increasing intracellular cAMP (79). This mechanism is mediated by the parasite circumsporozoite protein, and is inhibited by the blockade of CD91 or the removal of surface proteoglycans on KCs (79). By altering intracellular levels of cAMP, sporozoites are also thought to inhibit inflammatory cytokine production by KCs, instead favoring IL-10 secretion (80). Additionally, the phagocytic capacity of murine KCs towards bacteria has been shown to be substantially impaired during Plasmodium infection (81), further highlighting the compromising effects of malarial infection on KCs. The notion that KCs might exhibit pathogenic features during malarial infection is further supported by the finding that osteopetrotic (op/op) mice, which show a severe deficiency of mononuclear phagocytes including KCs, exhibit reduced hepatic infection (82). Paradoxically, depletion of KCs by clodronate liposomes augments parasitemia and hepatic infection (78, 82). Notably, clodronate liposome-mediated depletion of KCs is thought to compromise the structural integrity of the hepatic sinusoids, particularly by inducing gaps in the sinusoidal wall (82). Although this has been speculated to be the reason for the contradiction between the infection outcome following the aforementioned methods of KC-depletion, the authors provide no data to indicate that the integrity of the sinusoidal lining is unaltered in op/op mice (82).
Moreover, in a murine model of self-resolving Plasmodium infection invoked by the injection of parasitized erythrocytes, KCs are partially lost and replaced by infiltrating monocytes that in turn gave rise to long-lasting moKCs (83). In this model, KC loss occurs prior to the peak of blood parasitemia and coincides with an increase in the infiltration of monocytic cells (83). Notably, in vitro work has also suggested a role for ferroptosis and the deposition of hemozoin (81), a crystallized by-product of hemoglobin catabolism that is released in the circulation during malarial infection (84), as a trigger of KC death during Plasmodium infection (81). It is worth mentioning that a common feature in the pathology of many parasitic infections, including those invoked by Plasmodium and Trypanosoma, is the occurrence of anemia. In such settings, erythrocytes exhibit surface alterations resulting in enhanced erythro-phagocytosis by phagocytic cells (85, 86). As they are implicated in the clearance of damaged erythrocytes (24, 25), the erythro-phagocytic functions of KCs might also contribute to their response to parasitic infections. In this regard, erythro-phagocytosis has been shown to imprint an anti-inflammatory signature on KCs that is manifested in a lower expression of Ifng, Tnf, Il12b, Cxcl9 and Cxcl10 and MHCII-related genes in a mouse model of hemolytic anemia (87). Whether reprogramming due to erythro-phagocytosis functionally contributes to the response to parasitic infections is unclear.
During schistosomiasis, the main pathology results from the host type 2 immune response to the parasite eggs deposited in the liver. The eggs become surrounded by a granulomatous reaction encompassing mononuclear cells, eosinophils, neutrophils and fibroblasts, ultimately resulting in fibrosis (88). Alternatively activated macrophages, including KCs and monocyte-derived macrophages, are implicated in mediating the type 2 immune response and pathology in this context. Murine Schistosoma mansoni infection leads to the accumulation of Ly6Chi monocytes in the liver, which proliferate and differentiate into CD11bhi macrophages that gradually become the predominant hepatic macrophage population during the infection (89). KCs are steadily ablated as the infection proceeds, and although they showed little, if any, proliferative activity, they remain mainly of embryonic origin up to 10 weeks post-infection. Nevertheless, at this time-point, a minor but significant bone marrow contribution to the KC pool was observed (89). CD11bhi macrophages are alternatively activated in the infected livers and exhibit an IL4Ra-dependent upregulation of Ym1 and Relm-α. Although granuloma size was increased upon the deletion of Il4ra in myeloid cells (Lyz2Cre), this did not alter the survival kinetics (89). A recent scRNA-seq of Schistosoma japonica-infected murine livers also provided insights into macrophage heterogeneity during schistosomiasis (90). This study reported the presence of two major macrophage populations in the infected liver, a KC population (Timd4, Cd163, Marco), and a Ccr2+ population that lacked the expression of KC markers, and might hence represent infiltrating monocyte-derived macrophages (90). Noteworthy, the two populations were enriched in Chil3 expression, although KCs were notably more enriched in Arg1 and Retnla, suggesting a general alternative activation of macrophages in this context (90). Moreover, hepatic macrophages from S. mansoni-infected mice produce IL-6, IL-10, IL-4 and IL-13 in response to stimulation with soluble worm antigens and are thought to present worm antigens to hepatic T cells, promoting their type 2 differentiation (91). KCs are also implicated in tapeworm infection, particularly in cystic echinococcosis, as they uptake, via CLEC4F, the mucins of the laminated layer that protect the larval stages (hydatids) of Echinococcus granulosus parasites (92).
Overall, while KCs mount protective responses in some types of parasitic infections, they seem to be overwhelmed and even hijacked in other cases of parasitic insults. Notably, most experimental work investigating the roles of KCs in parasitic infection models has been done using non-specific KC targeting and identification methods that complicates the interpretation of the results. Hence, the recent advances in KC targeting and identification techniques might help resolve ambiguous aspects of KC biology during parasitic infections.
Perspectives: infection history and reprogramming of Kupffer cell function
As detailed previously, KCs are highly implicated in the response to pathogens. Given the wide range of pathogens that directly or indirectly target the liver, KC-pathogen interactions are probably a recurrent phenomenon during infections. In this regard, an increasing number of studies have shown that exposure to infections can alter tissue macrophage properties in the long-term. This effect can be mediated by ontogenic shifts, transcriptional and epigenetic reprogramming as well as alteration of the macrophage microenvironment (93–95). It is therefore likely that KCs are similarly subject to such modulation and that their functional properties are continuously shaped by infection history. For instance, although monocytes replenish KCs during almost all types of infectious diseases, it is unknown whether moKCs generated under these inflammatory scenarios are functionally distinct from emKCs. Although several studies report that moKCs generated following artificial KC-depletion during steady-state adopt the emKC identity and share some functional properties (7, 11, 17), this might not necessarily apply to situations where moKCs are generated in an inflammatory context. In support of this notion, it has been shown that monocyte-derived alveolar macrophages (moAMs), generated during influenza infection, are transcriptionally and epigenetically distinct from their embryonic counterparts and exhibit superior antibacterial functions (94). However, these enhanced antibacterial properties fail to develop when moAMs are generated following clodronate liposome-induced depletion of alveolar macrophages (94), confirming the need of inflammation to confer distinctive properties on monocyte-derived macrophages. In line with this, studies have highlighted some functional differences between emKCs and the moKCs generated during inflammatory contexts (17, 96). For instance, irradiation-generated moKCs are clearly superior to emKCs in the phagocytosis of a number of bacterial pathogens in vivo (17). In contrast, moKCs generated under lipemic conditions appear inferior to emKCs in their capacity to load and process lipids (96). Likewise, emKCs are more efficient in scavenging acetylated low density lipoprotein than irradiation-generated moKCs (17), indicating differences in the context of lipid metabolism. Moreover, emKCs are relatively radio-resistant as they upregulate the expression of Cdkna1 to resist and survive irradiation, whereas moKCs seem to lack this property (19). Importantly, although some of the distinctions between moKCs and emKCs might readily be made on the basis of the transcriptome, some may be represented in forms of “hidden” alterations at the level of the epigenome. In this context, despite several weeks of intrahepatic residency after irradiation, moKCs only recover less than half of the tissue-specific enhancer regions found in emKCs (14), further supporting the idea that ontogeny or the timing of moKC generation associates with differential functionality.
In addition to ontogeny-related effects, pathogens may directly alter macrophages, or their microenvironment. For instance, exposure to pathogens or their components is well documented to drive a persistent remodelling of the epigenomic landscapes of macrophages (95, 97). For instance, peripheral LPS administration can reprogram murine microglia and associates with altered responses to neurological disease (97). Likewise, exposure to inflammatory signals commonly encountered during infections, such as interferon-γ (paradigm of type 1 inflammation) or IL-4 (paradigm of type 2 inflammation), reshapes the macrophage epigenome and triggers the generation of latent enhancers associated with faster and more robust responses upon secondary stimulation (98, 99). Although this concept is rarely studied in KCs, a recent study has shown that in vitro treatment of KCs with β-glucan induces trained immunity, promoting an anti-inflammatory response to a secondary stimulation with LPS (61), confirming that KCs are amenable to reprogramming. Tissue macrophages, including KCs, rely on local signals provided by stromal cells in their tissue of residence in order to maintain their identity (2, 100–103). It is therefore logical that changes to the KC niche as a result of infections might subsequently reprogram KC identity and functions. This concept is exemplified in the lung, where changes in the microenvironment resulting from a previous exposure to experimental pneumonia can epigenetically reprogram embryonically-derived alveolar macrophages, leading to long-term impairment of their phagocytic function (95). In conclusion, despite the fact that KCs frequently and dynamically respond to infections, studies exploring whether a past exposure to infectious inflammation alters the KC phenotype are largely lacking. Major questions include whether infection history alters KC biology, and if so, for how long that lasts, and whether such effects are reversible. Importantly, what is the physiological consequence of such changes on the overall response to secondary inflammation? Given the frequent exposure of humans to infections and the important roles played by liver macrophages during infections, this topic warrants further investigation.
Author contributions
The manuscript was written by MM and revised by PB, KM and JG. All authors contributed to the article and approved the submitted version.
Funding
MM, KM and JG are supported by FWO (MM: 1157718N). KM holds an ERC Consolidator Grant.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity (2022) 55(9):1515–29. doi: 10.1016/j.immuni.2022.08.002
2. Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity (2019) 51(4):638–54.e9. doi: 10.1016/j.immuni.2019.08.017
3. Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science (2012) 336(6077):86. doi: 10.1126/science.1219179
4. Hoeffel G, Chen J, Lavin Y, Low D, Almeida Francisca F, See P, et al. C-myb+ Erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42(4):665–78. doi: 10.1016/j.immuni.2015.03.011
5. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518(7540):547–51. doi: 10.1038/nature13989
6. Sheng J, Ruedl C, Karjalainen K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity (2015) 43(2):382–93. doi: 10.1016/j.immuni.2015.07.016
7. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated kupffer cells. Nat Commun (2016) 7(1):10321. doi: 10.1038/ncomms10321
8. Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science (2016) 353(6304):aaf4238. doi: 10.1126/science.aaf4238
9. Scott CL, T’Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor zeb2 is required to maintain the tissue-specific identities of macrophages. Immunity (2018) 49(2):312–25.e5. doi: 10.1016/j.immuni.2018.07.004
10. Hagemeyer N, Kierdorf K, Frenzel K, Xue J, Ringelhan M, Abdullah Z, et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for irf8 in macrophage maturation. EMBO J (2016) 35(16):1730–44. doi: 10.15252/embj.201693801
11. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity (2019) 51(4):655–70.e8. doi: 10.1016/j.immuni.2019.09.002
12. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell (2022) 185(2):379–96.e38. doi: 10.1016/j.cell.2021.12.018
13. Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F, et al. Alk1 signaling is required for the homeostasis of kupffer cells and prevention of bacterial infection. J Clin Invest (2022) 132(3). doi: 10.1172/JCI150489
14. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
15. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13(11):1118–28. doi: 10.1038/ni.2419
16. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity (2020) 53(3):641–57.e14. doi: 10.1016/j.immuni.2020.08.004
17. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol (2016) 65(4):758–68. doi: 10.1016/j.jhep.2016.05.037
18. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in nash. Cell Rep (2021) 34(2). doi: 10.1016/j.celrep.2020.108626
19. Soysa R, Lampert S, Yuen S, Douglass AN, Li W, Pfeffer K, et al. Fetal origin confers radioresistance on liver macrophages via P21cip1/waf1. J Hepatol (2019) 71(3):553–62. doi: 10.1016/j.jhep.2019.04.015
20. Knoll P, Schlaak J, Uhrig A, Kempf P, zum Büschenfelde K-HM, Gerken G. Human kupffer cells secrete il-10 in response to lipopolysaccharide (Lps) challenge. J Hepatol (1995) 22(2):226–9. doi: 10.1016/0168-8278(95)80433-1
21. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatol (Baltimore Md) (2015) 62(1):279–91. doi: 10.1002/hep.27793
22. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic kupffer cells. Hepatol (Baltimore Md) (2008) 48(3):978–90. doi: 10.1002/hep.22395
23. Nairz M, Theurl I, Swirski FK, Weiss G. "Pumping iron"-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch (2017) 469(3-4):397–418. doi: 10.1007/s00424-017-1944-8
24. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med (2016) 22(8):945–51. doi: 10.1038/nm.4146
25. Terpstra V, van Berkel TJC. Scavenger receptors on liver kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood (2000) 95(6):2157–63. doi: 10.1182/blood.V95.6.2157
26. Theurl M, Theurl I, Hochegger K, Obrist P, Subramaniam N, van Rooijen N, et al. Kupffer cells modulate iron homeostasis in mice via regulation of hepcidin expression. J Mol Med (2008) 86(7):825. doi: 10.1007/s00109-008-0346-y
27. Lou D-Q, Lesbordes J-C, Nicolas G, Viatte L, Bennoun M, Van Rooijen N, et al. Iron- and inflammation-induced hepcidin gene expression in mice is not mediated by kupffer cells in vivo. Hepatol (Baltimore Md) (2005) 41(5):1056–64. doi: 10.1002/hep.20663
28. Li Y, Fu J, Ling Y, Yago T, McDaniel JM, Song J, et al. Sialylation on O-glycans protects platelets from clearance by liver kupffer cells. Proc Natl Acad Sci (2017) 114(31):8360. doi: 10.1073/pnas.1707662114
29. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES, et al. Macrophage galactose lectin is critical for kupffer cells to clear aged platelets. J Exp Med (2020) 217(4). doi: 10.1084/jem.20190723
30. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity (2017) 47(5):913–27.e6. doi: 10.1016/j.immuni.2017.10.006
31. Shi J, Fujieda H, Kokubo Y, Wake K. Apoptosis of neutrophils and their elimination by kupffer cells in rat liver. Hepatol (Baltimore Md) (1996) 24(5):1256–63. doi: 10.1002/hep.510240545
32. Shi J, Gilbert GE, Kokubo Y, Ohashi T. Role of the liver in regulating numbers of circulating neutrophils. Blood (2001) 98(4):1226–30. doi: 10.1182/blood.V98.4.1226
33. Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol (2018) 330:27–42. doi: 10.1016/j.cellimm.2018.01.020
34. Demetz E, Tymoszuk P, Hilbe R, Volani C, Haschka D, Heim C, et al. The haemochromatosis gene hfe and kupffer cells control ldl cholesterol homeostasis and impact on atherosclerosis development. Eur Heart J (2020) 41(40):3949–59. doi: 10.1093/eurheartj/ehaa140
35. Keshvari S, Caruso M, Teakle N, Batoon L, Sehgal A, Patkar OL, et al. Csf1r-dependent macrophages control postnatal somatic growth and organ maturation. PloS Genet (2021) 17(6):e1009605. doi: 10.1371/journal.pgen.1009605
36. Gao H, Jin Z, Bandyopadhyay G, Cunha e Rocha K, Liu X, Zhao H, et al. Mir-690 treatment causes decreased fibrosis and steatosis and restores specific kupffer cell functions in Nash. Cell Metab (2022) 34(7):978–90.e4. doi: 10.1016/j.cmet.2022.05.008
37. Blériot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. A subset of kupffer cells regulates metabolism through the expression of cd36. Immunity (2021) 54(9):2101–16.e6. doi: 10.1016/j.immuni.2021.08.006
38. Hume DA, Offermanns S, Bonnavion R. Contamination of isolated mouse kupffer cells with liver sinusoidal endothelial cells. Immunity (2022) 55(7):1139–40. doi: 10.1016/j.immuni.2022.06.010
39. Iannacone M, Blériot C, Andreata F, Ficht X, Laura C, Garcia-Manteiga JM, et al. Response to contamination of isolated mouse kupffer cells with liver sinusoidal endothelial cells. Immunity (2022) 55(7):1141–2. doi: 10.1016/j.immuni.2022.06.012
40. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity (2017) 47(2):374–88.e6. doi: 10.1016/j.immuni.2017.07.018
41. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature (2019) 572(7768):199–204. doi: 10.1038/s41586-019-1373-2
42. MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, et al. Single cell rna sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun (2018) 9(1):4383. doi: 10.1038/s41467-018-06318-7
43. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575(7783):512–8. doi: 10.1038/s41586-019-1631-3
44. Balmer ML, Slack E, de Gottardi A, Lawson MAE, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Trans Med (2014) 6(237):237ra66–ra66. doi: 10.1126/scitranslmed.3008618
45. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of Mass Cytometry and Imaging Analysis Reveals origin, Location, and Functional Repopulation Of liver myeloid Cells in Mice. Gastroenterology (2016) 151(6):1176–91. doi: 10.1053/j.gastro.2016.08.024
46. Lee W-Y, Moriarty TJ, Wong CHY, Zhou H, Strieter RM, van Rooijen N, et al. An intravascular immune response to borrelia burgdorferi involves kupffer cells and inkt cells. Nat Immunol (2010) 11(4):295–302. doi: 10.1038/ni.1855
47. Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, et al. Commensal-driven immune zonation of the liver promotes host defence. Nature (2021) 589(7840):131–6. doi: 10.1038/s41586-020-2977-2
48. Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, Kubes P. Crig functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe (2016) 20(1):99–106. doi: 10.1016/j.chom.2016.06.002
49. Broadley Steven P, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe (2016) 20(1):36–48. doi: 10.1016/j.chom.2016.05.023
50. Helmy KY, Katschke KJ, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. Crig: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124(5):915–27. doi: 10.1016/j.cell.2005.12.039
51. Min C, Park J, Kim G, Moon H, Lee S-A, Kim D, et al. Tim-4 functions as a scavenger receptor for phagocytosis of exogenous particles. Cell Death Dis (2020) 11(7):561. doi: 10.1038/s41419-020-02773-7
52. Duan Y, Chu H, Brandl K, Jiang L, Zeng S, Meshgin N, et al. Crig on liver macrophages clears pathobionts and protects against alcoholic liver disease. Nat Commun (2021) 12(1):7172. doi: 10.1038/s41467-021-27385-3
53. Wong CHY, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol (2013) 14(8):785–92. doi: 10.1038/ni.2631
54. McDonald B, Zucoloto AZ, Yu I-L, Burkhard R, Brown K, Geuking MB, et al. Programing of an intravascular immune firewall by the gut microbiota protects against pathogen dissemination during infection. Cell Host Microbe (2020) 28(5):660–8.e4. doi: 10.1016/j.chom.2020.07.014
55. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42(1):145–58. doi: 10.1016/j.immuni.2014.12.020
56. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17(3):151. doi: 10.1038/nri.2016.147
57. Di Paolo Nelson C, Doronin K, Baldwin Lisa K, Papayannopoulou T, Shayakhmetov Dmitry M. The transcription factor irf3 triggers “Defensive suicide” necrosis in response to viral and bacterial pathogens. Cell Rep (2013) 3(6):1840–6. doi: 10.1016/j.celrep.2013.05.025
58. Triantafyllou E, Gudd CLC, Mawhin M-A, Husbyn HC, Trovato FM, Siggins MK, et al. Pd-1 blockade improves kupffer cell bacterial clearance in acute liver injury. J Clin Invest (2021) 131(4). doi: 10.1172/JCI140196
59. Deppermann C, Peiseler M, Zindel J, Zbytnuik L, Lee W-Y, Pasini E, et al. Tacrolimus impairs kupffer cell capacity to control bacteremia: why transplant recipients are susceptible to infection. Hepatol (Baltimore Md) (2021) 73(5):1967–84. doi: 10.1002/hep.31499
60. Komine S, Akiyama K, Warabi E, Oh S, Kuga K, Ishige K, et al. Exercise training enhances in vivo clearance of endotoxin and attenuates inflammatory responses by potentiating kupffer cell phagocytosis. Sci Rep (2017) 7(1):11977. doi: 10.1038/s41598-017-12358-8
61. Zhang H, Chen T, Ren J, Xia Y, Onuma A, Wang Y, et al. Pre-operative exercise therapy triggers anti-inflammatory trained immunity of kupffer cells through metabolic reprogramming. Nat Metab (2021) 3(6):843–58. doi: 10.1038/s42255-021-00402-x
62. Wong YC, Tay SS, McCaughan GW, Bowen DG, Bertolino P. Immune outcomes in the liver: is cd8 T cell fate determined by the environment? J Hepatol (2015) 63(4):1005–14. doi: 10.1016/j.jhep.2015.05.033
63. Li M, Sun R, Xu L, Yin W, Chen Y, Zheng X, et al. Kupffer cells support hepatitis B virus–mediated cd8+ T cell exhaustion via hepatitis B core antigen–tlr2 interactions in mice. J Immunol (2015) 195(7):3100–9. doi: 10.4049/jimmunol.1500839
64. Xu L, Yin W, Sun R, Wei H, Tian Z. Kupffer cell-derived il-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatol (Baltimore Md) (2014) 59(2):443–52. doi: 10.1002/hep.26668
65. Bénéchet AP, De Simone G, Di Lucia P, Cilenti F, Barbiera G, Le Bert N, et al. Dynamics and genomic landscape of cd8+ T cells undergoing hepatic priming. Nature (2019) 574(7777):200–5. doi: 10.1038/s41586-019-1620-6
66. Spolski R, Li P, Leonard WJ. Biology and regulation of il-2: from molecular mechanisms to human therapy. Nat Rev Immunol (2018) 18(10):648–59. doi: 10.1038/s41577-018-0046-y
67. De Simone G, Andreata F, Bleriot C, Fumagalli V, Laura C, Garcia-Manteiga JM, et al. Identification of a kupffer cell subset capable of reverting the T Cell dysfunction induced by hepatocellular priming. Immunity (2021) 54(9):2089–100.e8. doi: 10.1016/j.immuni.2021.05.005
68. Wu LL, Peng WH, Wu HL, Miaw SC, Yeh SH, Yang HC, et al. Lymphocyte antigen 6 complex, locus C+ Monocytes and kupffer cells orchestrate liver immune responses against hepatitis B virus in mice. Hepatol (Baltimore Md) (2019) 69(6):2364–80. doi: 10.1002/hep.30510
69. Borst K, Frenz T, Spanier J, Tegtmeyer P-K, Chhatbar C, Skerra J, et al. Type I interferon receptor signaling delays kupffer cell replenishment during acute fulminant viral hepatitis. J Hepatol (2018) 68(4):682–90. doi: 10.1016/j.jhep.2017.11.029
70. Manickan E, Smith JS, Tian J, Eggerman TL, Lozier JN, Muller J, et al. Rapid kupffer cell death after intravenous injection of adenovirus vectors. Mol Ther (2006) 13(1):108–17. doi: 10.1016/j.ymthe.2005.08.007
71. He JQ, Katschke KJ Jr., Gribling P, Suto E, Lee WP, Diehl L, et al. Crig mediates early kupffer cell responses to adenovirus. J leukocyte Biol (2013) 93(2):301–6. doi: 10.1189/jlb.0612311
72. Liu G, Fu Y, Yosri M, Chen Y, Sun P, Xu J, et al. Crig plays an essential role in intravascular clearance of bloodborne parasites by interacting with complement. Proc Natl Acad Sci (2019) 116(48):24214–20. doi: 10.1073/pnas.1913443116
73. Liu G, Abas O, Strickland AB, Chen Y, Shi M. Cxcr6+Cd4+ T cells promote mortality during trypanosoma brucei infection. PloS Pathog (2021) 17(10):e1009968. doi: 10.1371/journal.ppat.1009968
74. Sun D, Sun P, Li H, Zhang M, Liu G, Strickland AB, et al. Fungal dissemination is limited by liver macrophage filtration of the blood. Nat Commun (2019) 10(1):4566. doi: 10.1038/s41467-019-12381-5
75. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol (2005) 5(9):722–35. doi: 10.1038/nri1686
76. Pradel G, Frevert U. Malaria sporozoites actively enter and pass through rat kupffer cells prior to hepatocyte invasion. Hepatol (Baltimore Md) (2001) 33(5):1154–65. doi: 10.1053/jhep.2001.24237
77. Frevert U, Engelmann S, Zougbédé S, Stange J, Ng B, Matuschewski K, et al. Intravital observation of plasmodium berghei sporozoite infection of the liver. PloS Biol (2005) 3(6):e192. doi: 10.1371/journal.pbio.0030192
78. Tavares J, Formaglio P, Thiberge S, Mordelet E, Van Rooijen N, Medvinsky A, et al. Role of host cell traversal by the malaria sporozoite during liver infection. J Exp Med (2013) 210(5):905–15. doi: 10.1084/jem.20121130
79. Usynin I, Klotz C, Frevert U. Malaria circumsporozoite protein inhibits the respiratory burst in kupffer cells. Cell Microbiol (2007) 9(11):2610–28. doi: 10.1111/j.1462-5822.2007.00982.x
80. Klotz C, Frevert U. Plasmodium yoelii sporozoites modulate cytokine profile and induce apoptosis in murine kupffer cells. Int J Parasitol (2008) 38(14):1639–50. doi: 10.1016/j.ijpara.2008.05.018
81. Hirako IC, Antunes MM, Rezende RM, Hojo-Souza NS, Figueiredo MM, Dias T, et al. Uptake of plasmodium chabaudi hemozoin drives kupffer cell death and fuels superinfections. Sci Rep (2022) 12(1):19805. doi: 10.1038/s41598-022-23858-7
82. Baer K, Roosevelt M, Clarkson AB Jr., Van Rooijen N, Schnieder T, Frevert U. Kupffer cells are obligatory for plasmodium yoelii sporozoite infection of the liver. Cell Microbiol (2007) 9(2):397–412. doi: 10.1111/j.1462-5822.2006.00798.x
83. Lai SM, Sheng J, Gupta P, Renia L, Duan K, Zolezzi F, et al. Organ-specific fate, recruitment, and refilling dynamics of tissue-resident macrophages during blood-stage malaria. Cell Rep (2018) 25(11):3099–109. doi: 10.1016/j.celrep.2018.11.059
84. Olivier M, Van Den Ham K, Shio M, Kassa F, Fougeray S. Malarial pigment hemozoin and the innate inflammatory response. Front Immunol (2014) 5. doi: 10.3389/fimmu.2014.00025
85. Stijlemans B, Cnops J, Naniima P, Vaast A, Bockstal V, De Baetselier P, et al. Development of a phrodo-based assay for the assessment of in vitro and in vivo erythrophagocytosis during experimental trypanosomosis. PloS Negl Trop Dis (2015) 9(3):e0003561. doi: 10.1371/journal.pntd.0003561
86. Waitumbi JN, Opollo MO, Muga RO, Misore AO, Stoute JA. Red cell surface changes and erythrophagocytosis in children with severe plasmodium falciparum anemia. Blood (2000) 95(4):1481–6. doi: 10.1182/blood.V95.4.1481.004k15_1481_1486
87. Pfefferlé M, Ingoglia G, Schaer CA, Yalamanoglu A, Buzzi R, Dubach IL, et al. Hemolysis transforms liver macrophages into antiinflammatory erythrophagocytes. J Clin Invest (2020) 130(10):5576–90. doi: 10.1172/JCI137282
88. Anthony BJ, Ramm GA, McManus DP. Role of resident liver cells in the pathogenesis of schistosomiasis. Trends Parasitol (2012) 28(12):572–9. doi: 10.1016/j.pt.2012.09.005
89. Rolot M, Dougall AM, Javaux J, Lallemand F, Machiels B, Martinive P, et al. Recruitment of hepatic macrophages from monocytes is independent of il-4rα but is associated with ablation of resident macrophages in schistosomiasis. Eur J Immunol (2019) 49(7):1067–81. doi: 10.1002/eji.201847796
90. Zhang Y, Li J, Li H, Jiang J, Guo C, Zhou C, et al. Single-cell rna sequencing to dissect the immunological network of liver fibrosis in schistosoma japonicum-infected mice. Front Immunol (2022) 13:980872. doi: 10.3389/fimmu.2022.980872
91. Hayashi N, Matsui K, Tsutsui H, Osada Y, Mohamed RT, Nakano H, et al. Kupffer cells from schistosoma mansoni-infected mice participate in the prompt type 2 differentiation of hepatic T cells in response to worm antigens1. J Immunol (1999) 163(12):6702–11. doi: 10.4049/jimmunol.163.12.6702
92. Barrios AA, Mouhape C, Schreiber L, Zhang L, Nell J, Suárez-Martins M, et al. Mucins shed from the laminated layer in cystic echinococcosis are captured by kupffer cells via the lectin receptor clec4f. Infection Immun (2023) 91(6):e00031–23. doi: 10.1128/iai.00031-23
93. Rua R, Lee JY, Silva AB, Swafford IS, Maric D, Johnson KR, et al. Infection drives meningeal engraftment by inflammatory monocytes that impairs cns immunity. Nat Immunol (2019) 20(4):407–19. doi: 10.1038/s41590-019-0344-y
94. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol (2020) 21(2):145–57. doi: 10.1038/s41590-019-0568-x
95. Roquilly A, Jacqueline C, Davieau M, Mollé A, Sadek A, Fourgeux C, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol (2020) 21(6):636–48. doi: 10.1038/s41590-020-0673-x
96. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity (2020) 53(3):627–40.e5. doi: 10.1016/j.immuni.2020.06.003
97. Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018) 556(7701):332–8. doi: 10.1038/s41586-018-0023-4
98. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell (2013) 152(1):157–71. doi: 10.1016/j.cell.2012.12.018
99. Kang K, Park SH, Chen J, Qiao Y, Giannopoulou E, Berg K, et al. Interferon-Γ Represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor maf. Immunity (2017) 47(2):235–50.e4. doi: 10.1016/j.immuni.2017.07.017
100. Bellomo A, Mondor I, Spinelli L, Lagueyrie M, Stewart BJ, Brouilly N, et al. Reticular fibroblasts expressing the transcription factor wt1 define a stromal niche that maintains and replenishes splenic red pulp macrophages. Immunity (2020) 53(1):127–42.e7. doi: 10.1016/j.immuni.2020.06.008
101. Buechler MB, Kim K-W, Onufer EJ, Williams JW, Little CC, Dominguez CX, et al. A stromal niche defined by expression of the transcription factor wt1 mediates programming and homeostasis of cavity-resident macrophages. Immunity (2019) 51(1):119–30.e5. doi: 10.1016/j.immuni.2019.05.010
102. Masuda T, Amann L, Monaco G, Sankowski R, Staszewski O, Krueger M, et al. Specification of cns macrophage subsets occurs postnatally in defined niches. Nature (2022) 604(7907):740–8. doi: 10.1038/s41586-022-04596-2
Keywords: liver, Kupffer cell, liver macrophages, infectious diseases, macrophage ontogeny
Citation: Musrati MA, De Baetselier P, Movahedi K and Van Ginderachter JA (2023) Ontogeny, functions and reprogramming of Kupffer cells upon infectious disease. Front. Immunol. 14:1238452. doi: 10.3389/fimmu.2023.1238452
Received: 11 June 2023; Accepted: 11 August 2023;
Published: 25 August 2023.
Edited by:
Thiago Almeida Pereira, Stanford University, United StatesReviewed by:
Zhaojun Wang, Shanghai Jiao Tong University, ChinaJanos G. Filep, Montreal University, Canada
Copyright © 2023 Musrati, De Baetselier, Movahedi and Van Ginderachter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jo A. Van Ginderachter, jo.van.ginderachter@vub.be
†These authors share senior authorship