
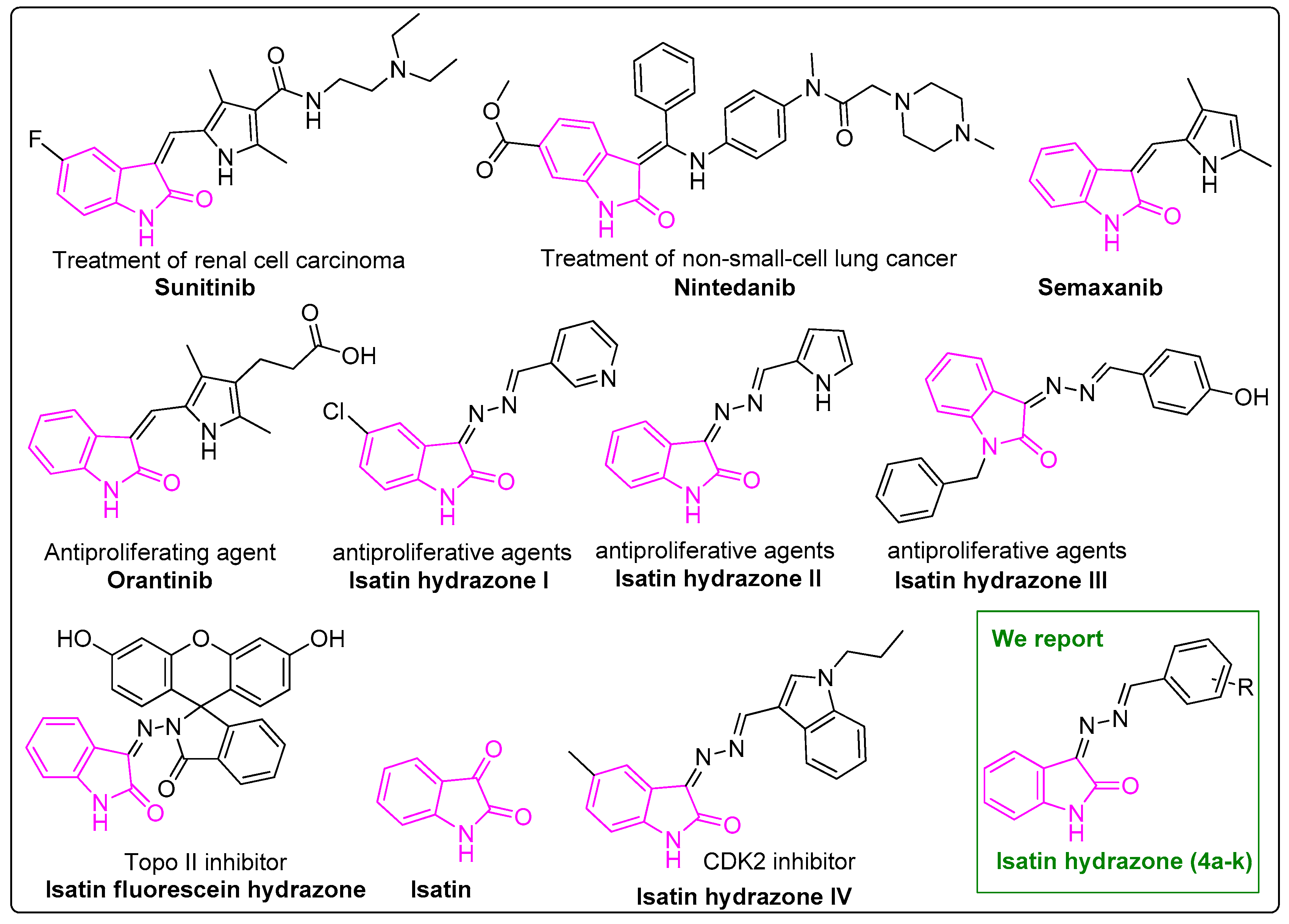
A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Isatin-Hydrazones (4)
2.2. Biological Evaluation
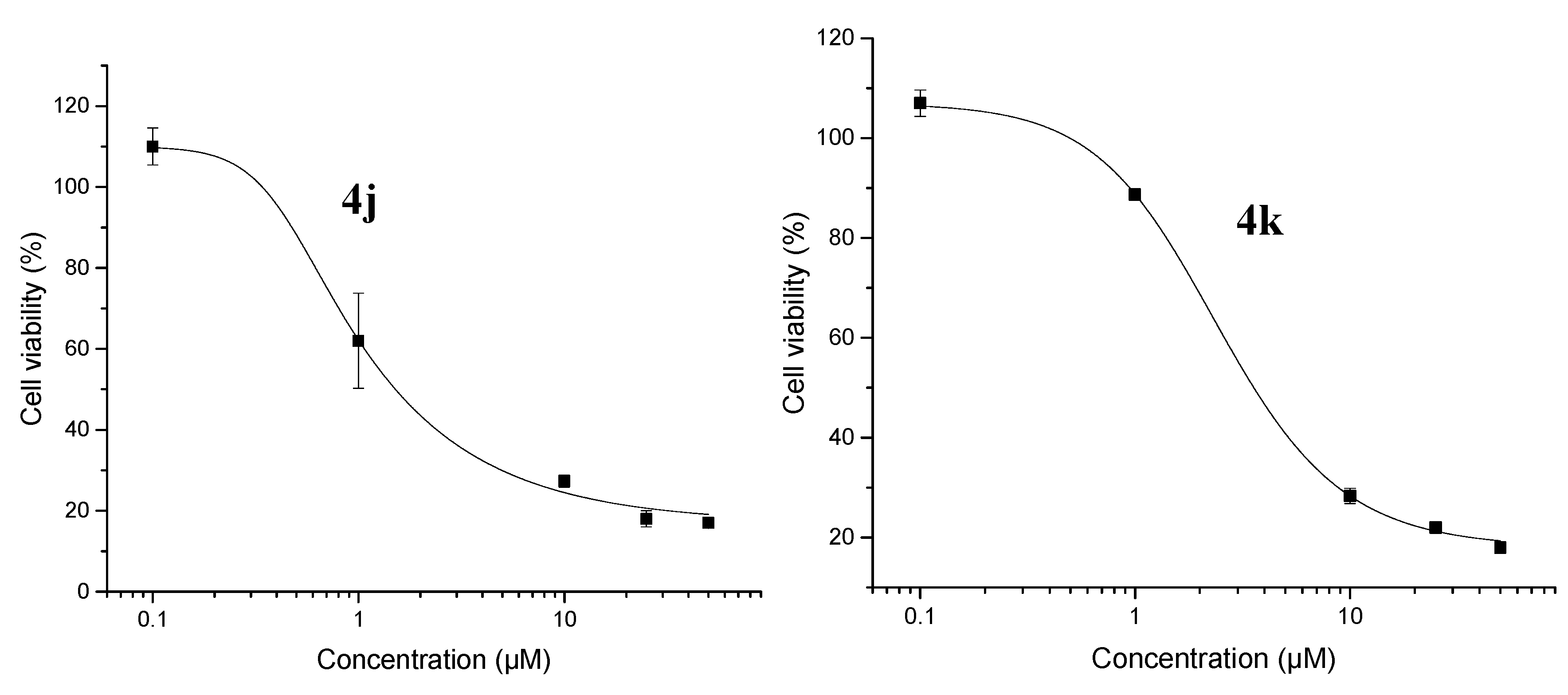
2.2.1. Cytotoxicity
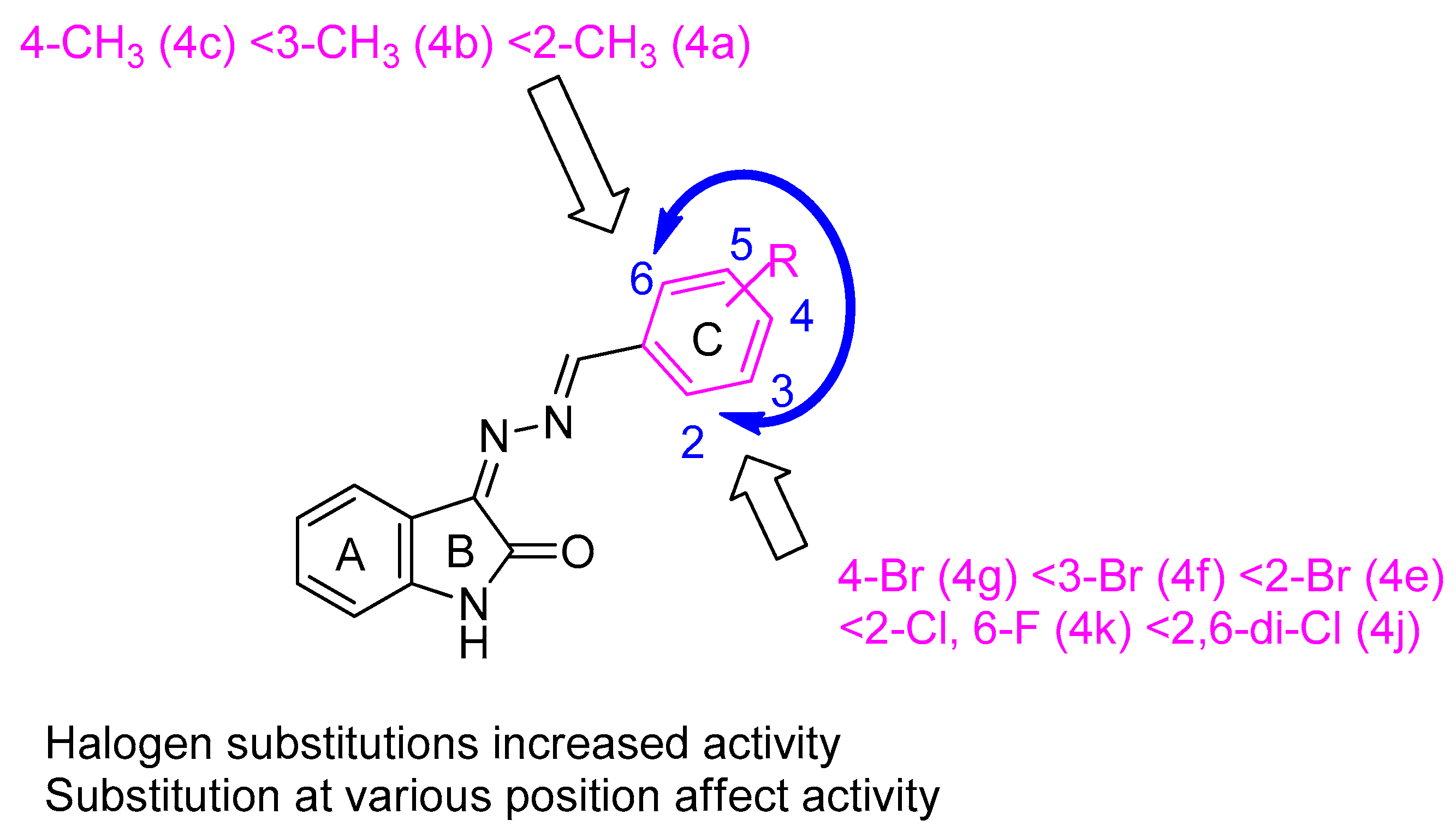
2.2.2. Structure–Activity Relationships (SARs) Study of 4a–k
2.2.3. CDK2 Protein Kinase Inhibitory Activity of 4a–k
2.3. In Silico Drug Likeness Property Analysis
2.4. Architecture of the CDK2 Active Site
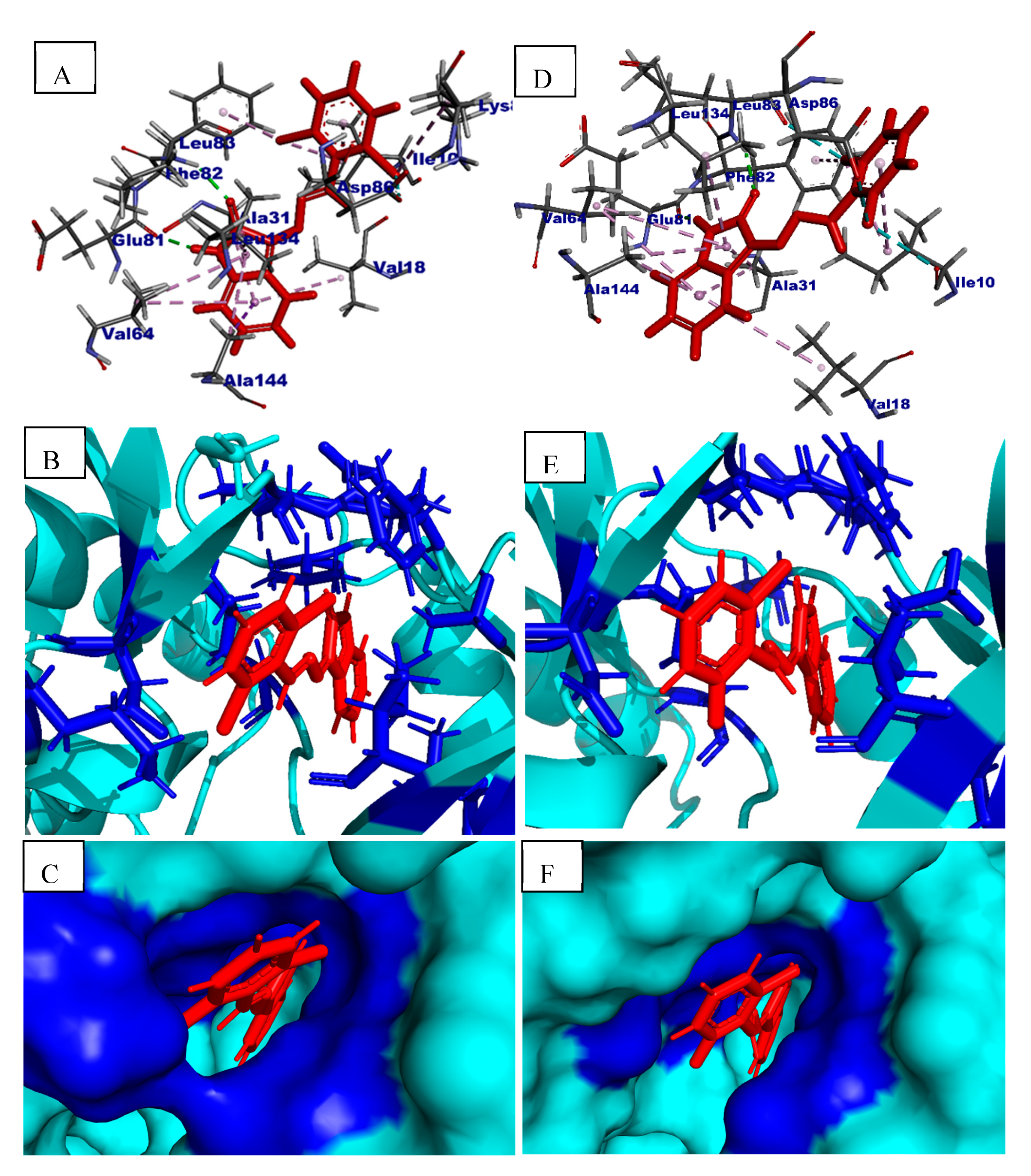
2.5. In Silico Binding Mechanism Analysis
3. Materials and Methods
3.1. General
3.2. (Z)-3-Hydrazonoindolin-2-one (2)
3.3. General Procedure for the Synthesis of 3-[benzylidene(substituted)hydrazono]indolin-2-ones 4a–k
3.3.1. 3-((2-Methylbenzylidene)hydrazono)indolin-2-one (4a)
3.3.2. 3-((3-Methylbenzylidene)hydrazono)indolin-2-one (4b)
3.3.3. 3-((4-Methylbenzylidene)hydrazono)indolin-2-one (4c)
3.3.4. 3-((4-(Methylthio)benzylidene)hydrazono)indolin-2-one (4d)
3.3.5. 3-((2-Bromobenzylidene)hydrazono)indolin-2-one (4e)
3.3.6. 3-((3-Bromobenzylidene)hydrazono)indolin-2-one (4f)
3.3.7. 3-((4-Bromobenzylidene)hydrazono)indolin-2-one (4g)
3.3.8. 3-((4-Methoxy-2,6-dimethylbenzylidene)hydrazono)indolin-2-one (4h)
3.3.9. 3-((2-Hydroxy-4-methoxybenzylidene)hydrazono)indolin-2-one (4i)
3.3.10. 3-((2,6-Dichlorobenzylidene)hydrazono)indolin-2-one (4j)
3.3.11. 3-((2-Chloro-6-fluorobenzylidene)hydrazono)indolin-2-one (4k)
3.4. Cytotoxicity
3.5. In Vitro Cyclin Dependent Kinase2 (CDK2) Inhibitory Activity
3.6. Molecular Docking and In-Silico ADME Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Control: Knowledge into ACTION. Available online: http://www.who.int/cancer/modules/en/ (accessed on 20 August 2020).
- Latest Global Cancer Data. Available online: https://www.iarc.fr/featured-news/latestglobal-cancer-data-cancer-burden-rises-to-18-1-million-new-casesand-9-6-million-cancer-deaths-in-2018/ (accessed on 20 August 2020).
- Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow/graphic-isotype?type=0&population=900&mode=population&sex=0&cancer=39&age_group=value&apc_male=0&apc_female=0 (accessed on 20 August 2020).
- Alsubi, T.A.; Attwa, M.W.; Bakheit, A.H.; Darwish, H.W.; Abuelizz, H.A.; Kadi, A.A. In silico and in vitro metabolism of ribociclib: A mass spectrometric approach to bioactivation pathway elucidation and metabolite profiling. RSC Adv. 2020, 10, 22668–22683. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Phase I metabolic profiling and unexpected reactive metabolites in human liver microsome incubations of X-376 using LC-MS/MS: Bioactivation pathway elucidation and in silico toxicity studies of its metabolites. RSC Adv. 2020, 10, 5412–5427. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Park, S.; Song, C.; Kadi, A.A.; Kwon, Y.; Rahman, A.F.M.M. Fluorescein hydrazones: A series of novel non-intercalative topoisomerase IIα catalytic inhibitors induce G1 arrest and apoptosis in breast and colon cancer cells. Eur. J. Med. Chem. 2017, 125, 49–67. [Google Scholar] [CrossRef]
- Rahman, A.F.M.M.; Park, S.-E.; Kadi, A.A.; Kwon, Y. Fluorescein Hydrazones as Novel Nonintercalative Topoisomerase Catalytic Inhibitors with Low DNA Toxicity. J. Med. Chem. 2014, 57, 9139–9151. [Google Scholar] [CrossRef]
- Ahmad, P.; Woo, H.; Jun, K.-Y.; Kadi, A.A.; Abdel-Aziz, H.A.; Kwon, Y.; Rahman, A.F.M.M. Design, synthesis, topoisomerase I & II inhibitory activity, antiproliferative activity, and structure–activity relationship study of pyrazoline derivatives: An ATP-competitive human topoisomerase IIα catalytic inhibitor. Bioorganic Med. Chem. 2016, 24, 1898–1908. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Abo-Ashour, M.F.; Almahli, H.; Alotaibi, O.J.; Al-Sanea, M.M.; Al-Ansary, G.H.; Ahmed, H.Y.; Elaasser, M.M.; Eldehna, W.M.; Abdel-Aziz, H.A. Novel [(N-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: Synthesis, biological evaluation and in silico studies. J. Enzym. Inhib. Med. Chem. 2020, 35, 1300–1309. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; El-Senduny, F.F.; Badria, F.A.; Rahman, A.F.M.M.; Barakat, A.; Elshaier, Y.A.M.M. Synthesis, Anticancer Activity, and Molecular Modeling of New Halogenated Spiro[pyrrolidine-thiazolo-oxindoles] Derivatives. Appl. Sci. 2020, 10, 2170. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, O.L. Untersuchungen über den Indigo. J. Prakt. Chem. 1840, 19, 321–362. [Google Scholar] [CrossRef]
- Laurent, A. Recherches sur l’indigo. Ann. Chim. Phys. 1840, 3, 40. [Google Scholar]
- Bergman, J.; Lindström, J.-O.; Tilstam, U. The structure and properties of some indolic constituents in Couroupita guianensis aubl. Tetrahedron 1985, 41, 2879–2881. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, F. TLC-UV spectrophotometric and TLC-scanning determination of isatin in leaf of Isatis. Zhong Cao Yao 1986, 17, 8–11. [Google Scholar]
- Yoshikawa, M.; Murakami, T.; Kishi, A.; Sakurama, T.; Matsuda, H.; Nomura, M.; Matsuda, H.; Kubo, M. Novel indoles, o-bisdesmoside, calanthoside, the precursor glycoside of tryptanthrin, indirubin, and isatin, with increasing skin blood flow promoting effects, from two calanthe species (orchidaceae). Chem. Pharm. Bull. 1998, 46, 886–888. [Google Scholar] [CrossRef]
- Popp, P.D. The Chemistry of Isatin. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Boulton, A.J., Eds.; Academic Press: London, UK, 1975; Volume 18, pp. 1–58. [Google Scholar]
- Silva, J.F.M.d.; Garden, S.J.; Pinto, A.C. The chemistry of isatins: A review from 1975 to 1999. J. Braz. Chem. Soc. 2001, 12, 273–324. [Google Scholar] [CrossRef]
- Sumpter, W.C. The Chemistry of Isatin. Chem. Rev. 1944, 34, 393–434. [Google Scholar] [CrossRef]
- Minami, M.; Hamaue, N.; Endo, T.; Hirafuji, M.; Terado, M.; Ide, H.; Yamazaki, N.; Yoshioka, M.; Ogata, A.; Tashiro, K. Effects of isatin, an endogenous MAO inhibitor, on dopamine (DA) and acetylcholine (ACh) concentrations in rats. Folia Pharmacol. Jpn. 1999, 114, 186–191. [Google Scholar] [CrossRef]
- Gillam, E.M.J.; Notley, L.M.; Cai, H.; De Voss, J.J.; Guengerich, F.P. Oxidation of Indole by Cytochrome P450 Enzymes. Biochemistry 2000, 39, 13817–13824. [Google Scholar] [CrossRef]
- Pervez, H.; Ahmad, M.; Zaib, S.; Yaqub, M.; Naseer, M.M.; Iqbal, J. Synthesis, cytotoxic and urease inhibitory activities of some novel isatin-derived bis-Schiff bases and their copper(II) complexes. MedChemComm 2016, 7, 914–923. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Abou-seri, S.M.; Ismail, N.S.M.; Elaasser, M.M.; Aly, M.H.; Abdel-Aziz, H.A. Bis-isatin hydrazones with novel linkers: Synthesis and biological evaluation as cytotoxic agents. Eur. J. Med. Chem. 2016, 108, 415–422. [Google Scholar] [CrossRef]
- Han, K.; Zhou, Y.; Liu, F.; Guo, Q.; Wang, P.; Yang, Y.; Song, B.; Liu, W.; Yao, Q.; Teng, Y.; et al. Design, synthesis and in vitro cytotoxicity evaluation of 5-(2-carboxyethenyl)isatin derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 591–594. [Google Scholar] [CrossRef]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Ranson, M.; Skropeta, D. Cytotoxic and anticancer activities of isatin and its derivatives: A comprehensive review from 2000–2008. Anti-Cancer Agents Med. Chem. 2009, 9, 397–414. [Google Scholar] [CrossRef]
- Matesic, L.; Locke, J.M.; Bremner, J.B.; Pyne, S.G.; Skropeta, D.; Ranson, M.; Vine, K.L. N-Phenethyl and N-naphthylmethyl isatins and analogues as in vitro cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 3118–3124. [Google Scholar] [CrossRef]
- Medvedev, A.; Igosheva, N.; Crumeyrolle-Arias, M.; Glover, V. Isatin: Role in stress and anxiety. Stress 2005, 8, 175–183. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Altoukhy, A.; Mahrous, H.; Abdel-Aziz, H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015, 90, 684–694. [Google Scholar] [CrossRef]
- Dweedar, H.E.; Mahrous, H.; Ibrahim, H.S.; Abdel-Aziz, H.A. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014, 78, 275–280. [Google Scholar] [CrossRef]
- Pakravan, P.; Kashanian, S.; Khodaei, M.M.; Harding, F.J. Biochemical and pharmacological characterization of isatin and its derivatives: From structure to activity. Pharmacol. Rep. PR 2013, 65, 313–335. [Google Scholar] [CrossRef]
- Medvedev, A.; Buneeva, O.; Gnedenko, O.; Ershov, P.; Ivanov, A. Isatin, an endogenous nonpeptide biofactor: A review of its molecular targets, mechanisms of actions, and their biomedical implications. BioFactors 2018, 44, 95–108. [Google Scholar] [CrossRef]
- Guo, H. Isatin derivatives and their anti-bacterial activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Pyne, S.G.; Bremner, J.B. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorganic Med. Chem. 2007, 15, 931–938. [Google Scholar] [CrossRef]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Pyne, S.G.; Bremner, J.B. An Investigation into the Cytotoxicity and Mode of Action of Some Novel N-Alkyl-Substituted Isatins. J. Med. Chem. 2007, 50, 5109–5117. [Google Scholar] [CrossRef]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Skropeta, D. Recent highlights in the development of isatin-based anticancer agents. Adv. Anticancer Agents Med. Chem. 2013, 2, 254–312. [Google Scholar]
- Nikalje, A.P.; Ansari, A.; Bari, S.; Ugale, V. Synthesis, Biological Activity, and Docking Study of Novel Isatin Coupled Thiazolidin-4-one Derivatives as Anticonvulsants. Archiv. Pharm. 2015, 348, 433–445. [Google Scholar] [CrossRef]
- Shingade, S.G.; Bari, S.B.; Waghmare, U.B. Synthesis and antimicrobial activity of 5-chloroindoline-2,3-dione derivatives. Med. Chem. Res. 2012, 21, 1302–1312. [Google Scholar] [CrossRef]
- Pandeya, S.; Smitha, S.; Jyoti, M.; Sridhar, S. Biological activities of isatin and its derivatives. Acta Pharm. 2005, 55, 27–46. [Google Scholar]
- Saha, S.; Acharya, C.; Pal, U.; Chowdhury, S.R.; Sarkar, K.; Maiti, N.C.; Jaisankar, P.; Majumder, H.K. A Novel Spirooxindole Derivative Inhibits the Growth of Leishmania donovani Parasites both In Vitro and In Vivo by Targeting Type IB Topoisomerase. Antimicrob. Agents Ch. 2016, 60, 6281–6293. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, S.; Debnath, B. Molecular docking studies and ADME prediction of novel isatin analogs with potent anti-EGFR activity. Med. Chem. 2014, 4, 558–568. [Google Scholar] [CrossRef]
- Singh, A.; Raghuwanshi, K.; Patel, V.K.; Jain, D.K.; Veerasamy, R.; Dixit, A.; Rajak, H. Assessment of 5-substituted Isatin as Surface Recognition Group: Design, Synthesis, and Antiproliferative Evaluation of Hydroxamates as Novel Histone Deacetylase Inhibitors. Pharm. Chem. J. 2017, 51, 366–374. [Google Scholar] [CrossRef]
- Varun; Sonam; Kakkar, R. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. MedChemComm 2019, 10, 351–368. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abou-Seri, S.M.; Supuran, C.T. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur. J. Med. Chem. 2018, 157, 28–36. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; Gratteri, P.; Eissa, I.H.; Fares, M.; Ismael, O.E.; Ghabbour, H.A.; Elaasser, M.M.; Abdel-Aziz, H.A.; et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur. J. Med. Chem. 2017, 139, 250–262. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Fares, M.; Ceruso, M.; Ghabbour, H.A.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; Abou El Ella, D.A.; Supuran, C.T. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur. J. Med. Chem. 2016, 110, 259–266. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, A.; Padhani, A.; Hayes, C.; Kakkar, A.J.; Leach, M.; Trigo, J.M.; Scurr, M.; Raynaud, F.; Phillips, S.; Aherne, W.; et al. A Phase I study of the angiogenesis inhibitor SU5416 (semaxanib) in solid tumours, incorporating dynamic contrast MR pharmacodynamic end points. Br. J. Cancer 2005, 93, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.M.; Feldman, D.R.; Ginsberg, M.S.; Kroog, G.; Tickoo, S.K.; Jia, X.; Georges, M.; Patil, S.; Baum, M.S.; Reuter, V.E.; et al. Phase II trial of sunitinib in patients with metastatic non-clear cell renal cell carcinoma. Investig. New Drugs 2012, 30, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, P.M.; Wolff, R.A.; Bogaard, K.; Waldrum, S.; Abbruzzese, J.L. A Phase I study of escalating doses of the tyrosine kinase inhibitor semaxanib (SU5416) in combination with irinotecan in patients with advanced colorectal carcinoma. Jpn. J. Clin. Oncol. 2006, 36, 100–103. [Google Scholar] [CrossRef]
- Marko, D.; Schätzle, S.; Friedel, A.; Genzlinger, A.; Zankl, H.; Meijer, L.; Eisenbrand, G. Inhibition of cyclin-dependent kinase 1 (CDK1) by indirubin derivatives in human tumour cells. Br. J. Cancer 2001, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramson, H.N.; Corona, J.; Davis, S.T.; Dickerson, S.H.; Edelstein, M.; Frye, S.V.; Gampe, R.T.; Harris, P.A.; Hassell, A.; Holmes, W.D.; et al. Oxindole-Based Inhibitors of Cyclin-Dependent Kinase 2 (CDK2): Design, Synthesis, Enzymatic Activities, and X-ray Crystallographic Analysis. J. Med. Chem. 2001, 44, 4339–4358. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.; Crumeyrolle-Arias, M.; Cardona, A.; Sandler, M.; Glover, V. Natriuretic peptide interaction with [3H]isatin binding sites in rat brain. Brain Res. 2005, 1042, 119–124. [Google Scholar] [CrossRef]
- Lawrence, H.R.; Pireddu, R.; Chen, L.; Luo, Y.; Sung, S.-S.; Szymanski, A.M.; Yip, M.L.R.; Guida, W.C.; Sebti, S.M.; Wu, J.; et al. Inhibitors of Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (Shp2) Based on Oxindole Scaffolds. J. Med. Chem. 2008, 51, 4948–4956. [Google Scholar] [CrossRef] [Green Version]
- Al-Salem, H.S.; Abuelizz, H.A.; Issa, I.S.; Mahmoud, A.Z.; AlHoshani, A.; Arifuzzaman, M.; Rahman, A.F.M.M. Synthesis of Novel Potent Biologically Active N-Benzylisatin-Aryl Hydrazones in Comparison with Lung Cancer Drug ‘Gefitinib’. Appl. Sci. 2020, 10, 3669. [Google Scholar] [CrossRef]
- Ekins, S.; Waller, C.L.; Swaan, P.W.; Cruciani, G.; Wrighton, S.A.; Wikel, J.H. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. Methods 2000, 44, 251–272. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Chemi, G.; Gemma, S.; Campiani, G.; Brogi, S.; Butini, S.; Brindisi, M. Computational Tool for Fast in silico Evaluation of hERG K+ Channel Affinity. Front. Chem. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Han, Y.; Hopfinger, A.J. Predicting Caco-2 Cell Permeation Coefficients of Organic Molecules Using Membrane-Interaction QSAR Analysis. J. Chem. Inf. Comput. Sci. 2002, 42, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Talapati, S.R.; Nataraj, V.; Pothuganti, M.; Gore, S.; Ramachandra, M.; Antony, T.; More, S.S.; Krishnamurthy, N.R. Structure of cyclin-dependent kinase 2 (CDK2) in complex with the specific and potent inhibitor CVT-313. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 350–356. [Google Scholar] [CrossRef]
- Sabt, A.; Eldehna, W.M.; Al-Warhi, T.; Alotaibi, O.J.; Elaasser, M.M.; Suliman, H.; Abdel-Aziz, H.A. Discovery of 3,6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: Synthesis, biological evaluation and in silico insights. J. Enzym. Inhib. Med. Chem. 2020, 35, 1616–1630. [Google Scholar] [CrossRef]
- Mohammad, T.; Batra, S.; Dahiya, R.; Baig, M.H.; Rather, I.A.; Dong, J.-J.; Hassan, I. Identification of high-affinity inhibitors of cyclin-dependent kinase 2 towards anticancer therapy. Molecules 2019, 24, 4589. [Google Scholar] [CrossRef] [Green Version]
- Coffey, K.E.; Moreira, R.; Abbas, F.Z.; Murphy, G.K. Synthesis of 3,3-dichloroindolin-2-ones from isatin-3-hydrazones and (dichloroiodo)benzene. Org. Biomol. Chem. 2015, 13, 682–685. [Google Scholar] [CrossRef]
- Afsah, E.M.; Elmorsy, S.S.; Abdelmageed, S.M.; Zaki, Z.E. Synthesis of some new mixed azines, Schiff and Mannich bases of pharmaceutical interest related to isatin. Z. Nat. B J. Chem. Sci. 2015, 70, 393–402. [Google Scholar] [CrossRef]
- Bkhaitan, M.M.; Mirza, A.Z.; Abdalla, A.N.; Shamshad, H.; Ul-Haq, Z.; Alarjah, M.; Piperno, A. Reprofiling of full-length phosphonated carbocyclic 2′-oxa-3′-aza-nucleosides toward antiproliferative agents: Synthesis, antiproliferative activity, and molecular docking study. Chem. Biol. Drug Des. 2017, 90, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arifuzzaman, M.; Hamza, A.; Zannat, S.S.; Fahad, R.; Rahman, A.; Hosen, S.M.Z.; Dash, R.; Hossain, M.K. Targeting galectin-3 by natural glycosides: A computational approach. Netw. Modeling Anal. Health Inform. Bioinform. 2020, 9, 14. [Google Scholar] [CrossRef]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins Struct. Funct. Bioinform. 2014, 82, 1646–1655. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 4a–k are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | |
|---|---|---|
| MCF7 | A2780 | |
| 4a | 10.82 ± 0.05 | >50 |
| 4b | 14 ± 1.33 | >50 |
| 4c | 32.48 ± 0.52 | >50 |
| 4d | 24 ± 2.61 | >50 |
| 4e | 5.46 ± 0.71 | 19 ± 2.52 |
| 4f | 9.07 ± 0.59 | 25 ± 2.82 |
| 4g | 15.70 ± 0.78 | >50 |
| 4h | 25.78 ± 0.13 | >50 |
| 4i | 7.77 ± 0.008 | >50 |
| 4j | 1.51 ± 0.09 | 26 ± 2.24 |
| 4k | 3.56 ± 0.31 | 27 ± 3.20 |
| Doxorubicin | 3.10 ± 0.29 | 0.20 ± 0.03 |
| Compounds | CDK2 Protein Kinase (IC50 in µM) * |
|---|---|
| 4j | 0.2456 |
| 4k | 0.3006 |
| Imatinib | 0.1312 |
| No. | MW a | HBD b | HBA c | logPo/w d | logS e | logP | HERG f | Caco-2 g | BBB h | MDCK i | HOA(%) j |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 263 | 1 | 5 | 2.59 | −3.82 | 9.8 | −5.84 | 1324 | −0.49 | 670 | 100 |
| 4b | 263 | 1 | 5 | 2.62 | −3.95 | 9.7 | −5.89 | 1317 | −0.51 | 666 | 100 |
| 4c | 263 | 1 | 5 | 2.62 | −3.95 | 9.7 | −5.89 | 1317 | −0.51 | 666 | 100 |
| 4d | 295 | 1 | 5.5 | 2.93 | −4.31 | 10.0 | -6.03 | 1310 | −0.49 | 1126 | 100 |
| 4e | 328 | 1 | 5 | 2.83 | −4.06 | 9.9 | −5.90 | 1323 | −0.33 | 1584 | 100 |
| 4f | 328 | 1 | 5 | 2.88 | −4.19 | 9.8 | −5.91 | 1317 | −0.32 | 1766 | 100 |
| 4g | 328 | 1 | 5 | 2.88 | −4.19 | 9.8 | −5.91 | 1317 | −0.32 | 1766 | 100 |
| 4h | 307 | 1 | 5.75 | 2.89 | −4.14 | 9.8 | −5.58 | 1760 | −0.44 | 911 | 100 |
| 4i | 295 | 2 | 6.5 | 1.79 | −3.39 | 12.4 | −5.74 | 487 | −1.07 | 227 | 86 |
| 4j | 318 | 1 | 5 | 3.12 | −4.35 | 9.6 | −5.67 | 1616 | −0.13 | 3162 | 100 |
| 4k | 302 | 1 | 5 | 2.90 | −4.17 | 9.7 | −5.75 | 1432 | −0.24 | 2158 | 100 |
| Doxo k | 544 | 5 | 15 | -0.49 | −2.37 | 24.2 | −6.02 | 2.29 | −2.95 | 0.766 | 0 |
| Compounds | Docking Score | Interacting Residues | Types of Interaction |
|---|---|---|---|
| 4j | −6.5 | Ile10, Val18, Ala31, Val64, Glu81, Phe82, Leu83, Asp86, Lys89, Leu134 and Ala144 | Hydrogen π-Alkyl Halogen π-σ |
| 4k | −5.9 | Ile10, Val18, Ala31, Val64, Glu81, Phe82, Leu83, Asp86, Leu134 and Ala144 | Hydrogen σ-Alkyl Halogen |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Salem, H.S.; Arifuzzaman, M.; Alkahtani, H.M.; Abdalla, A.N.; Issa, I.S.; Alqathama, A.; Albalawi, F.S.; Rahman, A.F.M.M. A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor. Molecules 2020, 25, 4400. https://doi.org/10.3390/molecules25194400
Al-Salem HS, Arifuzzaman M, Alkahtani HM, Abdalla AN, Issa IS, Alqathama A, Albalawi FS, Rahman AFMM. A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor. Molecules. 2020; 25(19):4400. https://doi.org/10.3390/molecules25194400
Chicago/Turabian StyleAl-Salem, Huda S., Md Arifuzzaman, Hamad M. Alkahtani, Ashraf N. Abdalla, Iman S. Issa, Aljawharah Alqathama, Fatemah S. Albalawi, and A. F. M. Motiur Rahman. 2020. "A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor" Molecules 25, no. 19: 4400. https://doi.org/10.3390/molecules25194400