Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
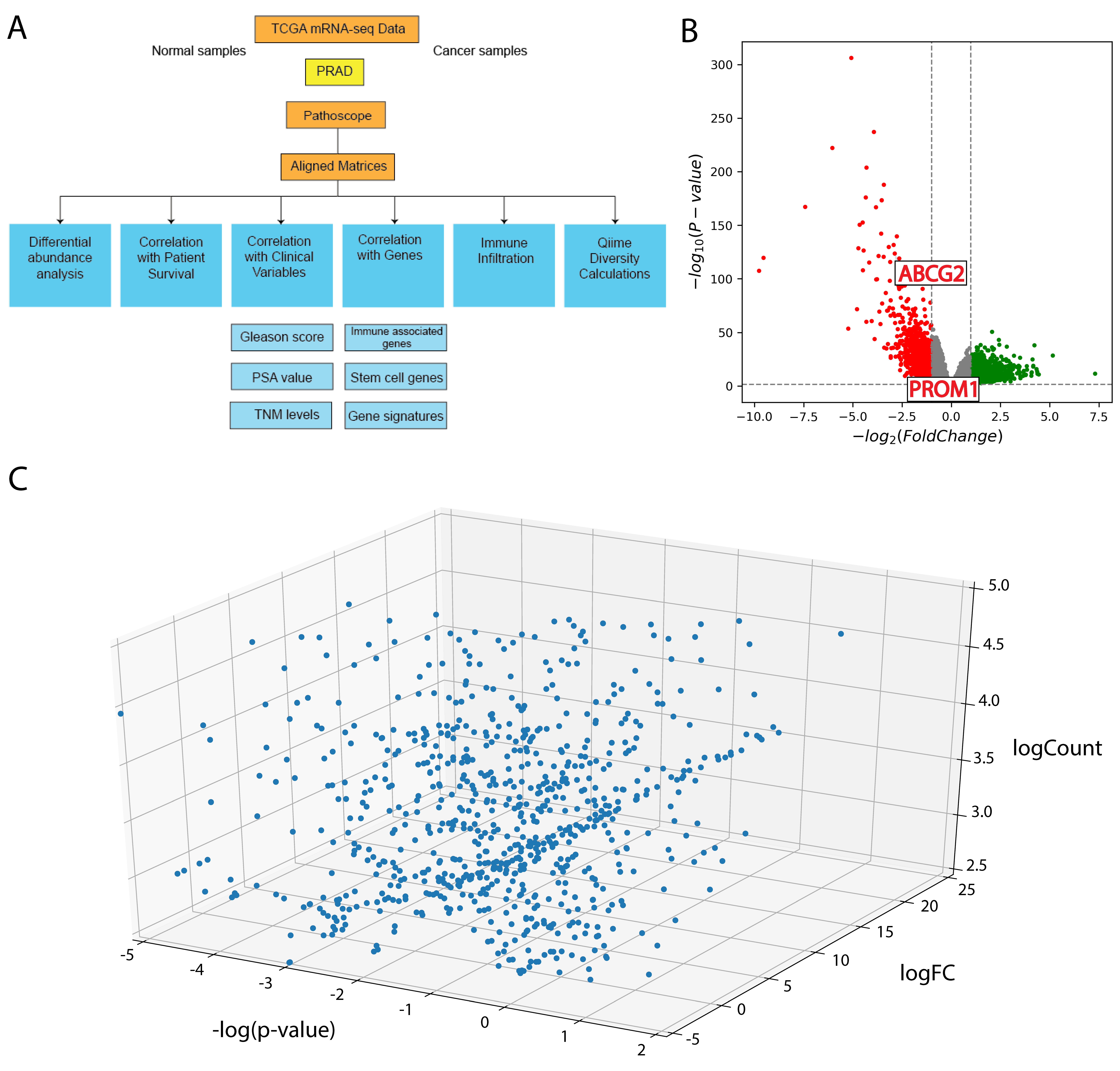
2.1. Data Acquisition
2.2. Differential Expression of Genes and Differential Abundance of Microbes
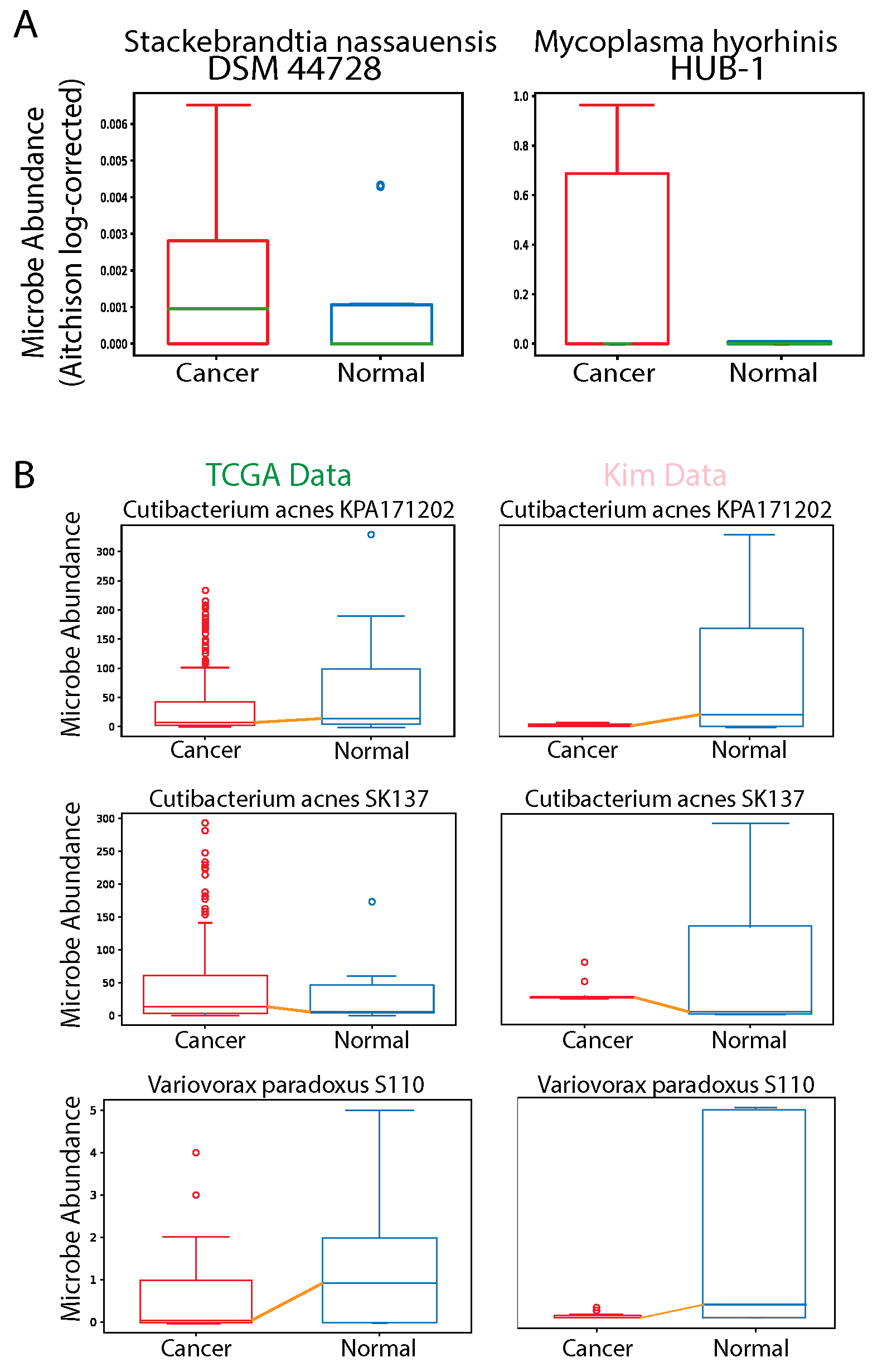
2.3. Comparison between Microbial Abundance of Tumor Samples and Available Adjacent Normal Samples
2.4. Contamination Correction
2.5. Microbe Correlation with Clinical Variables
2.5.1. Gleason Scores
2.5.2. TNM Stages
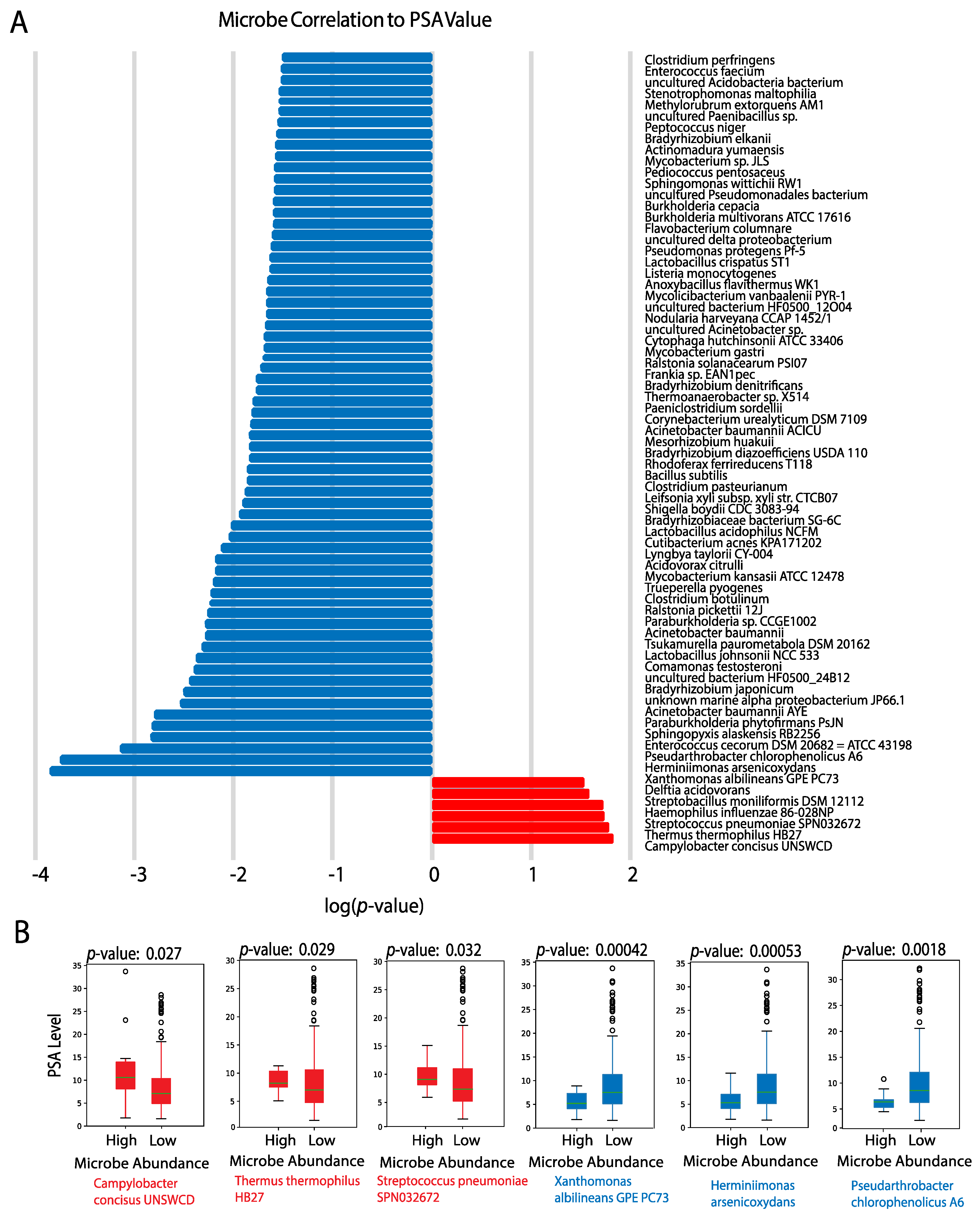
2.5.3. PSA Values
2.6. Correlation to Immune-Associated Processes
2.6.1. Correlation to Genomic Alterations (REVEALER)
2.6.2. Correlation to Immune-Associated (IA) Genes
2.6.3. Correlation to Immune Cell Types
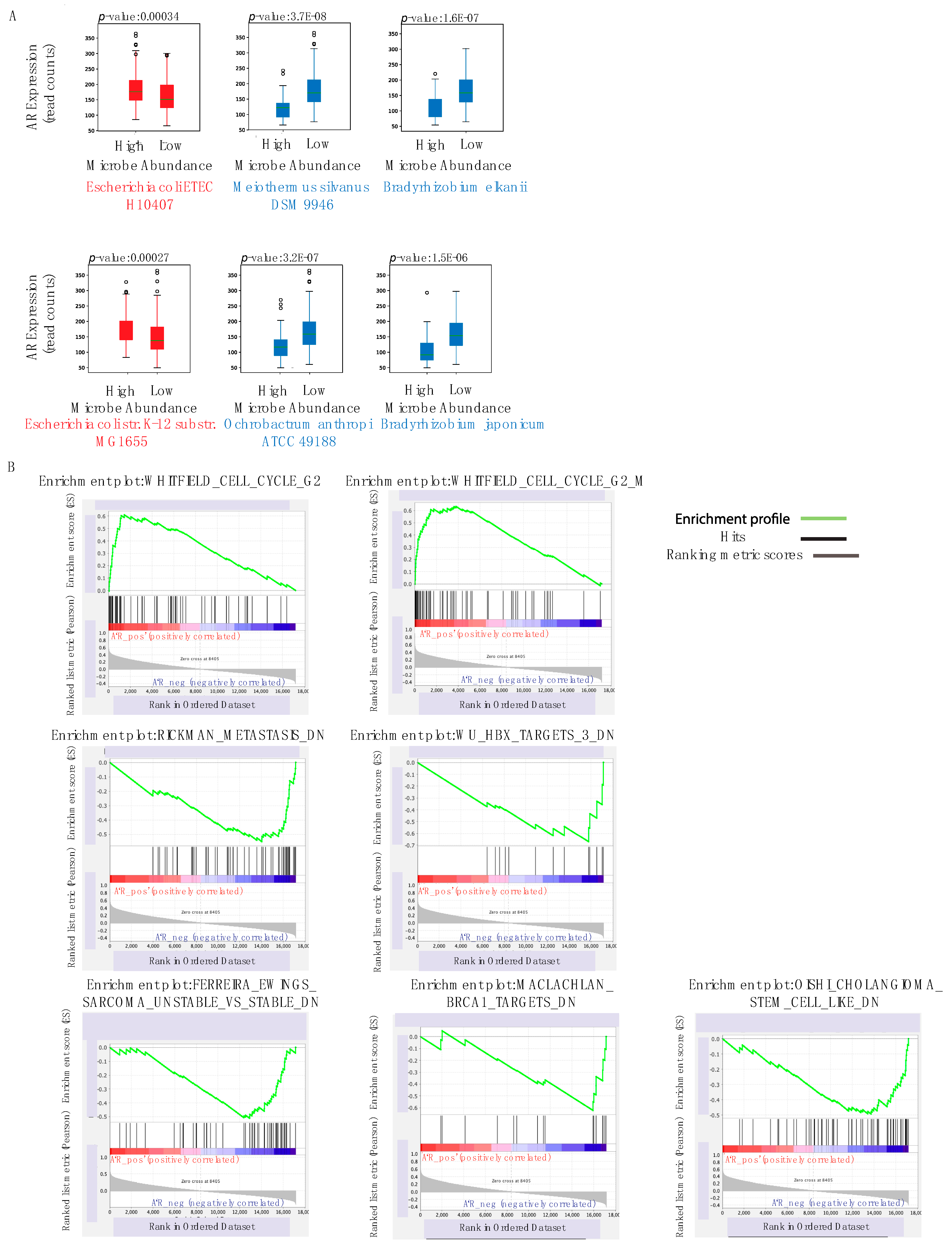
2.7. Correlation to AR Expression
2.8. Correlation to Stem-Cell Genes
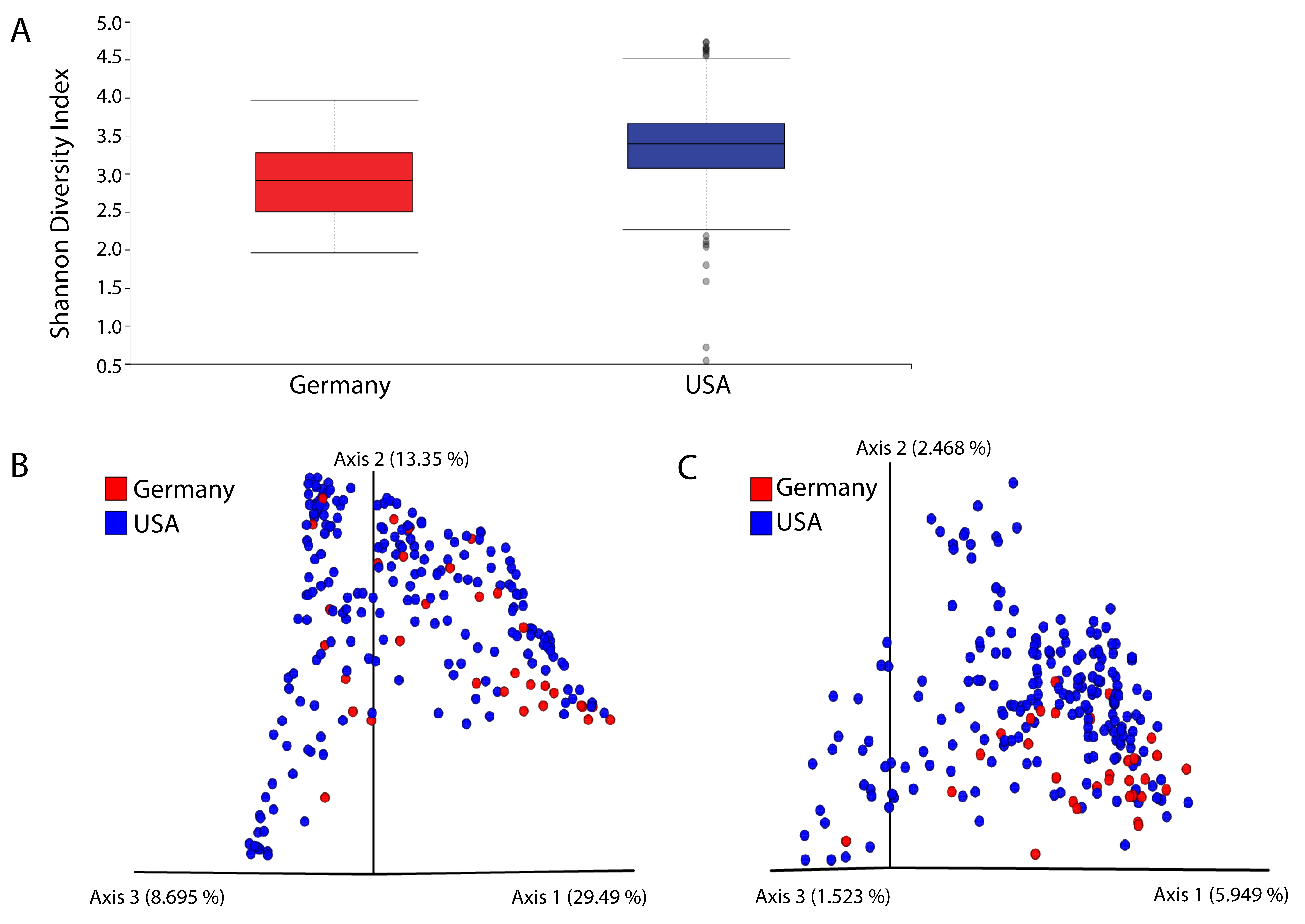
2.9. Qiime Diversity Calculations
2.10. Validation with Kim Dataset
3. Discussion
4. Materials and Methods
4.1. Data Acquisition from TCGA
4.2. Differential Microbial Abundance between Cancer and Normal Patients
4.3. Evaluation of Contamination Using Plates, Flow Cells, and Date of Sequencing
4.3.1. Read Counts from Sequencing
4.3.2. Aligning Microbe Abundance by Date Sequenced
4.4. Correlation of Microbial Abundance to Clinical Variables
4.5. Probability of Correlation Overlap
4.6. Correlation of Microbial Abundance with Genomic Alterations Using REVEALER
4.7. Correlation of Microbial Abundance with IA Gene Expression
4.8. Correlation of Microbial Abundance to Immune Cell Types
4.9. Correlation of Microbial Abundance to Expression of Immune-Associated Genes, AR, and Stem-Cell Genes
4.10. Correlation of Microbial Abundance with Immune Pathway Using GSEA
4.11. Qiime2 Diversity Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Na, L. Cancer Facts & Figures; American Cancer Society: Atlanta, GR, USA, 2020. [Google Scholar]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursell, L.K.; Metcalf, J.; Wegner Parfrey, L.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70 (Suppl. S1), S38–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The Contributions of prostate cancer stem cells in prostate cancer initiation and metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The human microbiome and cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.Y.; Shaw, M.H.; Redondo, G.; Nunez, G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008, 68, 10060–10067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, W.S. The gut microbiota and colon cancer. Science 2019, 364, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, G.; Jansson, J.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Erdman, S.E.; Poutahidis, T. The microbiome modulates the tumor macroenvironment. Oncoimmunology 2014, 3, e28271. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, D.N.; Dickson, R.P.; Moore, B.B. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J. Immunol. 2016, 196, 4839–4847. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 2019, 178, 795–806. [Google Scholar] [CrossRef]
- Urbaniak, C.; Cummins, J.; Brackstone, M.; Macklaim, J.; Gloor, G.B.; Baban, C.K.; Scott, L.; O’Hanlon, D.M.; Burton, J.P.; Francis, K.P.; et al. Microbiota of human breast tissue. Appl. Environ. Microbiol. 2014, 80, 3001–3007. [Google Scholar] [CrossRef] [Green Version]
- Thomas-White, K.; Brady, M.; Wolfe, A.J.; Mueller, E.R. The bladder is not sterile: History and current discoveries on the urinary microbiome. Curr. Bladder Dysfunct. Rep. 2016, 11, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Chi, C.; Fan, L.; Dong, B.; Shao, X.; Xie, S.; Li, M.; Xue, W. The microbiome of prostate fluid is associated with prostate cancer. Front. Microbiol. 2019, 10, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Gail, M.H.; Consonni, D.; Carugano, M.; Humphrys, M.; Pesatori, A.C.; Caporaso, N.E.; Goedert, J.J.; Ravel, J.; Landi, M.T. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genom. Biol. 2016, 17, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucevic Popovic, V.; Situm, M.; Chow, C.E.T.; Chan, L.S.; Roje, B.; Terzic, J. The urinary microbiome associated with bladder cancer. Sci. Rep. 2018, 8, 812157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, C.M.; Shrestha, E.; Peiffer, L.B.; Sfanos, K.S. The microbiome in prostate inflammation and prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 345–354. [Google Scholar] [CrossRef]
- Cavarretta, I.; Ferrarese, R.; Cazzaniga, W.; Saita, D.; Luciano, R.; Ceresola, E.R.; Locatelli, I.; Visconti, L.; Lavorgna, G.; Briganti, A.; et al. The microbiome of the prostate tumor microenvironment. Eur. Urol. 2017, 72, 625–631. [Google Scholar] [CrossRef]
- Allen-Vercoe, E.; Jobin, C. Fusobacterium and Enterobacteriaceae: Important players for CRC? Immunol. Lett. 2014, 162 Pt A, 54–61. [Google Scholar] [CrossRef] [Green Version]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Gronberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Sfanos, K.S.; Yegnasubrabramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Markowski, M.C.; Peiffer, L.B.; Ernst, S.E.; White, J.R.; Pienta, K.J.; Antonarakis, E.S.; Ross, A.E. Compositional differences in gastrointestinal microbiota in prostate cancer patients treated with androgen axis-targeted therapies. Prostate Cancer Prostatic Dis. 2018, 21, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Davin-Regli, A.; Pages, J.M. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front Microbiol. 2015, 6, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilgin, H.; Sarmis, A.; Tigen, E.; Soyletir, G.; Mulazimoglu, L. Delftia acidovorans: A rare pathogen in immunocompetent and immunocompromised patients. Can. J. Infect. Dis. Med. Microbiol. 2015, 26, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Arora, N.C.; Sahai, K. Report on the newly emerging nosocomial Burkholderia cepacia in a tertiary hospital. Med. J. Armed Forces India 2016, 72 (Suppl. S1), S50–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, V.; Ghosh, A.R.; Bishayee, K.; Khuda-Bukhsh, A.R. Appraisal of the anti-cancer potential of probiotic Pediococcus pentosaceus GS4 against colon cancer: In vitro and in vivo approaches. J. Funct. Foods 2016, 23, 66–79. [Google Scholar] [CrossRef]
- Wood, L.M.; Guirnalda, P.D.; Seavey, M.M.; Paterson, Y. Cancer immunotherapy using Listeria monocytogenes and listerial virulence factors. Immunol. Res. 2008, 42, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Machata, S.; Tchatalbachev, S.; Mohamed, W.; Jänsch, L.; Hain, T.; Chakraborty, T. Lipoproteins of Listeria monocytogenes are critical for virulence and TLR2-mediated immune activation. J. Immunol. 2008, 181, 2028–2035. [Google Scholar] [CrossRef] [Green Version]
- Brunt, L.M.; Portnoy, D.A.; Unanue, E.R. Presentation of Listeria monocytogenes to CD8+ T cells requires secretion of hemolysin and intracellular bacterial growth. J. Immunol. 1990, 145, 3540–3546. [Google Scholar]
- Wood, L.M.; Paterson, Y. Attenuated Listeria monocytogenes: A powerful and versatile vector for the future of tumor immunotherapy. Front. Cell. Infect. Microbiol. 2014, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Castro, F.; Paterson, Y.; Gravekamp, C. High efficacy of a Listeria-based vaccine against metastatic breast cancer reveals a dual mode of action. Cancer Res. 2009, 69, 5860–5866. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, M.; Evangelista, M.; Lascio, N.D.; Kusmic, C.; Massa, A.; Orso, F.; Sarti, S.; Marranci, A.; Rodzik, K.; Germelli, L.; et al. Antitumoral effects of attenuated Listeria monocytogenes in a genetically engineered mouse model of melanoma. Oncogene 2019, 38, 3756–3762. [Google Scholar] [CrossRef]
- Motevaseli, E.; Khorramizadeh, M.R.; Hadjati, J.; Bonab, S.F.; Eslami, S.; Ghafouri-Fard, S. Investigation of antitumor effects of Lactobacillus crispatus in experimental model of breast cancer in BALB/c mice. Immunotherapy 2018, 10, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bagdure, S.R.; Fisher, M.A.; Ryan, M.E.; Khasawneh, F.A. Rhodococcus erythropolis encephalitis in patient receiving rituximab. Emerg. Infect. Dis. 2012, 18, 1377–1379. [Google Scholar] [CrossRef]
- Li, L.; Tarrand, J.J.; Han, X.Y. Microbiological and clinical features of four cases of catheter-related infection by Methylobacterium radiotolerans. J. Clin. Microbiol. 2015, 53, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Boktour, M.; Hanna, H.; Ansari, S.; Bahna, B.; Hachem, R.; Tarrand, J.; Rolston, K.; Safdar, A.; Raad, I. Central venous catheter and Stenotrophomonas maltophilia bacteremia in cancer patients. Cancer 2006, 106, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.T.; Chen, T.Y.; Su, W.C.; Yen, C.J.; Tsao, C.J. Implantable venous port-related infections in cancer patients. Support Care Cancer. 2004, 12, 197–201. [Google Scholar] [CrossRef]
- Safdar, A.; Rolston, K.V. Stenotrophomonas maltophilia: Changing spectrum of a serious bacterial pathogen in patients with cancer. Clin. Infect. Dis. 2007, 45, 1602–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazaki, A.A.; Pritsa, A.A.; Kyriakidis, D.A. Biotechnologically relevant enzymes from Thermus thermophilus. Appl. Microbiol. Biotechnol. 2002, 58, 1–12. [Google Scholar] [CrossRef]
- Pritsa, A.A.; Papazisis, K.T.; Kortsaris, A.H.; Geromichalos, G.D.; Kyriakidis, D.A. Antitumor activity of L-asparaginase from Thermus thermophilus. Anticancer Drugs. 2001, 12, 137–142. [Google Scholar] [CrossRef]
- Sircar, K.; Huang, H.; Hu, L.; Cogdell, D.; Dhillon, J.; Tzelepi, V.; Efstathiou, E.; Koumakpayi, I.H.; Saad, F.; Luo, D.; et al. Integrative molecular profiling reveals asparagine synthetase is a target in castration-resistant prostate cancer. Am. J. Pathol. 2012, 180, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Teng, J.L.; Botelho, M.G.; Lo, R.C.; Lau, S.K.; Woo, P.C. Arginine Metabolism in Bacterial Pathogenesis and Cancer Therapy. Int. J. Mol. Sci. 2016, 17, 363. [Google Scholar] [CrossRef] [Green Version]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 2017, 77, 6777–6787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, W.; Alrammah, H.; Kiernan, M.; Tonks, A.J.; Jackson, S.K. Lysophosphatidylcholine acyltransferase 2 (LPCAT2) co-localises with TLR4 and regulates macrophage inflammatory gene expression in response to LPS. Nat. Sci. Rep. 2020, 10, 10355. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; Isaacs, W.B.; De Marzo, A.M. Infections and inflammation in prostate cancer. Am. J. Clin. Exp. Urol. 2013, 1, 3–11. [Google Scholar] [PubMed]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002, 13, 1977–2000. [Google Scholar] [CrossRef]
- Ma, W.L.; Lai, H.C.; Yeh, S.; Cai, X.; Chang, C. Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocrinol. Relat. Cancer 2014, 21, R165–R182. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Papadopoulou, N.; Gehring, E.M.; Nasir, O.; Dimas, K.; Bhavsar, S.K.; Föller, M.; Alevizopoulos, K.; Lang, F.; Stournaras, C. Functional membrane androgen receptors in colon tumors trigger pro-apoptotic responses in vitro and reduce drastically tumor incidence in vivo. Mol. Cancer 2009, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Moltzahn, F.; Thalmann, G.N. Cancer stem cells in prostate cancer. Transl. Androl. Urol. 2013, 2, 242–253. [Google Scholar] [CrossRef]
- Hollern, D.P.; Swiatnicki, M.R.; Andrechek, E.R. Histological subtypes of mouse mammary tumors reveal conserved relationships to human cancers. PLoS Genet. 2018, 14, e1007135. [Google Scholar] [CrossRef]
- Rickman, D.S.; Millon, R.; De Reynies, A.; Thomas, E.; Wasylyk, C.; Muller, D.; Abecassis, J.; Wasylyk, B. Prediction of future metastasis and molecular characterization of head and neck squamous-cell carcinoma based on transcriptome and genome analysis by microarrays. Oncogene 2008, 27, 6607–6622. [Google Scholar] [CrossRef] [Green Version]
- Sokhansanj, B.A.; Rodrigue, G.R.; Fitch, J.P.; Wilson, D.M., III. A quantitative model of human DNA base excision repair. I. Mechanistic insights. Nucl. Acids Res. 2002, 30, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Dhanasekaran, S.; Prensner, J.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res. 2020, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabbagh, F.; Moradpour, Z.; Ghasemian, A.; Ghasemi, Y. Phylogeny of urate oxidase producing bacteria: On the basis of gene sequences of 16s rrna and uricase protein p37. Iran. J. Pharm. Sci. 2012, 8, 99–102. [Google Scholar]
- Gong, M.; Meng, L.; Jiang, B.; Zhang, J.; Yang, H.; Wu, J.; Shou, C. From mycoplasma hyorhinis promotes cancer cell invasiveness and metastasis through activation of MMP-2 and followed by phosphorylation of EGFR. Mol. Cancer Ther. 2008, 7, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLoS ONE 2013, 8, e77955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanek, C.; Goodison, S.; Chang, M.; Porvasnik, S.; Sakamoto, N.; Li, C.; Boehlein, S.; Rosser, C. Detection of antibodies directed at M. Hyorhinis p37 in the serum of men with newly diagnosed prostate cancer. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [Green Version]
- Brooke, J.S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.; Shende, N.; Li, W.; Castaneda, G.; Apostol, L.; Chang, E.Y.; Ongkeko, W.M. Comparative analysis of age-and gender-associated microbiome in lung adenocarcinoma and lung squamous cell carcinoma. Cancers 2020, 12, 1447. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Deshpande, N.P.; Wilkins, M.R.; Tan, C.G.; Burgos-Portugal, J.A.; Raftery, M.J.; Day, A.S.; Lemberg, D.A.; Mitchell, H. The pathogenic potential of Campylobacter concisus strains associated with chronic intestinal diseases. PLoS ONE 2011, 6, e29045. [Google Scholar] [CrossRef] [Green Version]
- Kaakoush, N.O.; Mitchell, H.M. Campylobacter concisus-A new player in intestinal disease. Front. Cell Infect. Microbiol. 2012, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Loughran, A.J.; Orihuela, C.J.; Tuomanen, E.I. Streptococcus pneumoniae. Invas. Inflamm. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Ullah, A.; Khakwani, Z.; Mehmood, H. Prostate abscess caused by community-acquired methicillin-resistant Staphylococcus aureus. J. Investig. Med. High Impact Case Rep. 2018, 6. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Troncoso, P.; Johnston, D.; Bucana, C.D.; Tahara, E.; Fidler, I.J.; Pettaway, C.A. Relative expression of type IV collagenase, E-cadherin, and vascular endothelial growth factor/vascular permeability factor in prostatectomy specimens distinguishes organ-confined from pathologically advanced prostate cancers. Clin. Cancer Res. 2000, 6, 2295–2308. [Google Scholar] [PubMed]
- Nnabugwu, I.; Udeh, E.; Ugwumba, F.; Ozoemena, F. Predicting Gleason score using the initial serum total prostate-specific antigen in Black men with symptomatic prostate adenocarcinoma in Nigeria. Clin. Interv. Aging 2016, 11, 961–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, K.; Ikeda, H.; Maolake, A.; Machioka, K.; Nohara, T.; Narimoto, K.; Ueno, S.; Kadono, Y.; Kitagawa, Y.; Konaka, H.; et al. The relationship between prostate-specific antigen and tnm classification or gleason score in prostate cancer patients with low prostate-specific antigen levels. Prostate 2015, 75, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Stephan, C.; Lein, M.; Jung, K.; Schnorr, D.; Loening, S. The influence of prostate volume on the ratio of free to total prostate specific antigen in serum of patients with prostate carcinoma and benign prostate hyperplasia. Cancer 1997, 79, 104–109. [Google Scholar] [CrossRef]
- Chen, L.; Davey Smith, G.; Evans, D.; Cox, A.; Lawlor, D.; Donovan, J.; Yuan, W.; Day, I.N.M.; Martin, R.M.; Lane, A.; et al. Genetic variants in the vitamin d receptor are associated with advanced prostate cancer at diagnosis: Findings from the prostate testing for cancer and treatment study and a systematic review. Cancer Epidemiol. Biomark. Prevent. 2009, 18, 2874–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asbell, S.O.; Raimane, K.C.; Montesano, A.T.; Zeitzer, K.L.; Asbell, M.D.; Vijayakumar, S. Prostate-specific antigen and androgens in African-American and white normal subjects and prostate cancer patients. J. Natl. Med. Assoc. 2000, 92, 445–449. [Google Scholar] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Gnanasekar, A.; Lee, A.; Li, W.T.; Haas, M.; Wang-Rodriguez, J.; Chang, E.Y.; Rajasekaran, M.; Ongkeko, W.M. Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer. Cancers 2020, 12, 2524. https://doi.org/10.3390/cancers12092524
Ma J, Gnanasekar A, Lee A, Li WT, Haas M, Wang-Rodriguez J, Chang EY, Rajasekaran M, Ongkeko WM. Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer. Cancers. 2020; 12(9):2524. https://doi.org/10.3390/cancers12092524
Chicago/Turabian StyleMa, Jiayan, Aditi Gnanasekar, Abby Lee, Wei Tse Li, Martin Haas, Jessica Wang-Rodriguez, Eric Y. Chang, Mahadevan Rajasekaran, and Weg M. Ongkeko. 2020. "Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer" Cancers 12, no. 9: 2524. https://doi.org/10.3390/cancers12092524