
Hypoxia-Driven HIF-1α Activation Reprograms Pre-Activated NK Cells towards Highly Potent Effector Phenotypes via ERK/STAT3 Pathways

 ,
,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
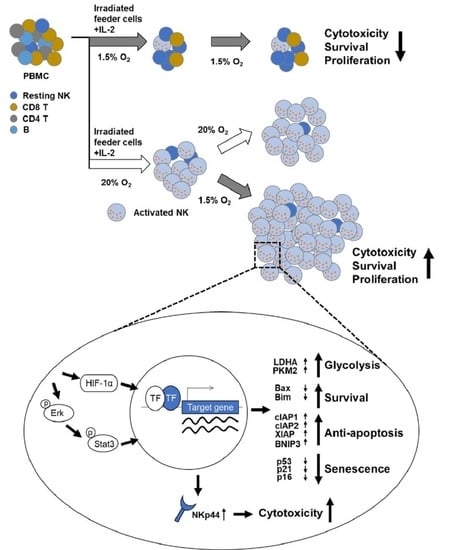
2. Results
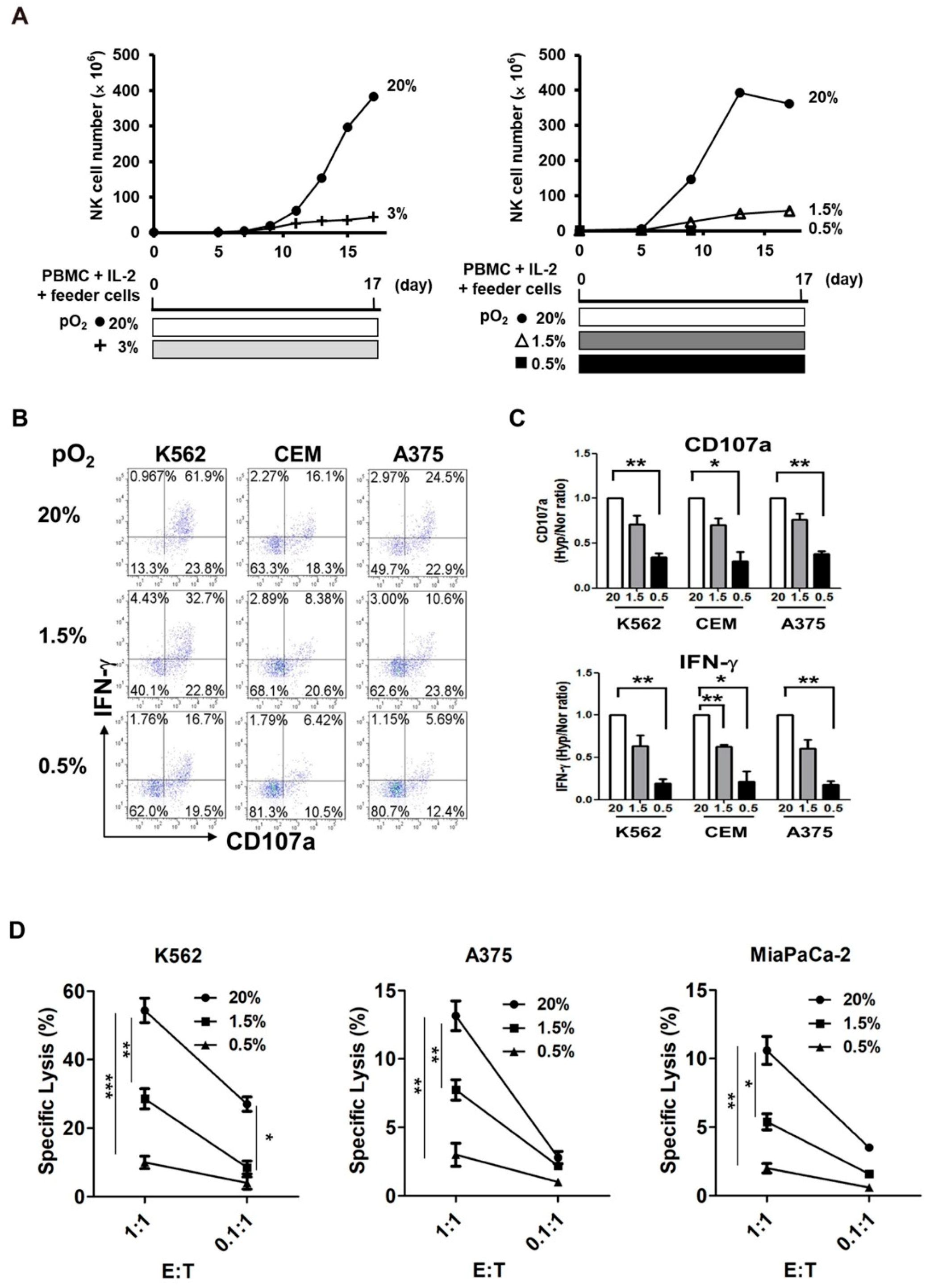
2.1. Exposure of NK Cells to Hypoxia Impairs Their Proliferation and Tumor Cytotoxicity
2.2. Pre-Activation of PBMCs Prior to Hypoxic Exposure Increases Ex-Vivo NK Cell Expansion
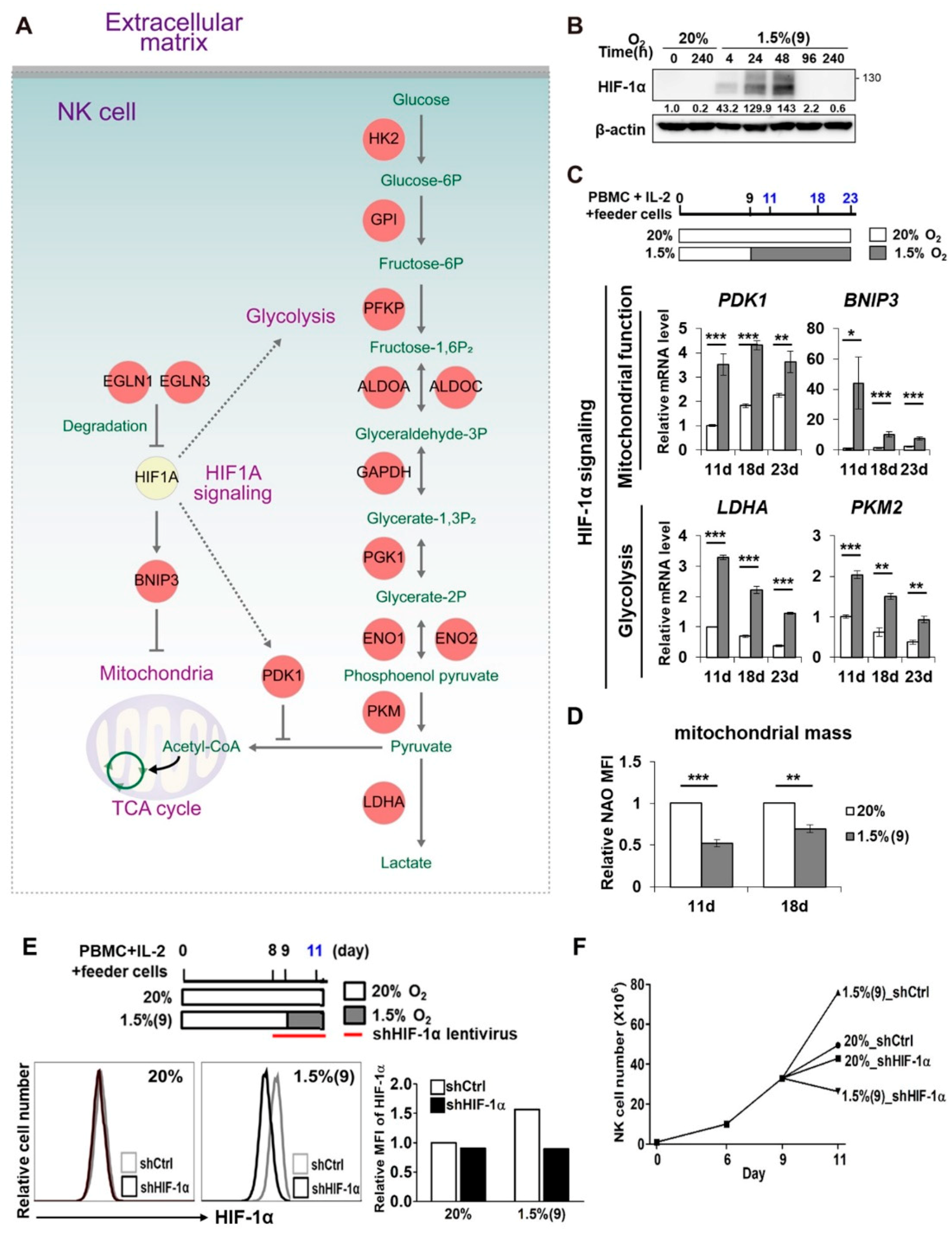
2.3. Pre-Activated NK Cells Show Downregulation of Genes Involved in Cell Cycle Arrest and Cell Death When Exposed to Hypoxia
2.4. HIF-1α Is Important for the Enhanced Proliferation of Pre-Activated Hypoxic NK Cells
2.5. Enhanced Proliferation of Pre-Activated Hypoxic NK Cells Is Mediated by ERK and STAT3 Signaling Pathways
2.6. Pre-Activated Hypoxic NK Cells Show Enhanced Cytotoxicity after Hypoxic Switch
2.7. Pre-Activated Hypoxic NK Cells Exhibit Higher NKp44 Expression Both at the Transcript and Protein Levels
3. Discussion
4. Materials and Methods
4.1. Enrichment and Expansion of NK Cells under Hypoxic Conditions
4.2. Cell Culture and Reagents
4.3. Flow Cytometry
4.4. Proliferation and Apoptosis Assay
4.5. 10-N Nonyl-Acridine Orange (NAO) Staining for Measuring Mitochondrial Mass
4.6. Cr Release Assay
4.7. Cytotoxicity Assay
4.8. CD107a Granule Release Assay and Intracellular Cytokine Staining
4.9. Transduction of NK Cells with Lentiviral Vectors Encoding shHIF-1α
4.10. In-Vivo Cytotoxicity Assay
4.11. Western Blot Analysis
4.12. Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.13. mRNA-Sequencing and Data Analysis
4.14. Identification of DEGs
4.15. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Kulkarni, N.; Shilpi; Lal, G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. OncoImmunology 2016, 5, e1235106. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548.e519. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front Immunol 2015, 6, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef]
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Kim, S.-J.; Lee, K.-M. NK Cell-Based Immunotherapies in Cancer. Immune Netw. 2020, 20. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef] [PubMed]
- Solocinski, K.; Padget, M.R.; Fabian, K.P.; Wolfson, B.; Cecchi, F.; Hembrough, T.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. Overcoming hypoxia-induced functional suppression of NK cells. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Nakayama, K.; Kataoka, N. Regulation of Gene Expression under Hypoxic Conditions. Int. J. Mol. Sci. 2019, 20, 3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Chan, A.T.; Meyerhardt, J.A.; Chung, D.C.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 2010, 176, 2292–2301. [Google Scholar] [CrossRef]
- Jögi, A.; Ehinger, A.; Hartman, L.; Alkner, S. Expression of HIF-1α is related to a poor prognosis and tamoxifen resistance in contralateral breast cancer. PLoS ONE 2019, 14, e0226150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birner, P.; Schindl, M.; Obermair, A.; Plank, C.; Breitenecker, G.; Oberhuber, G. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000, 60, 4693–4696. [Google Scholar]
- Liu, Q.; Cao, P. Clinical and prognostic significance of HIF-1α in glioma patients: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 22073–22083. [Google Scholar]
- Zhao, X.; Gao, S.; Ren, H.; Sun, W.; Zhang, H.; Sun, J.; Yang, S.; Hao, J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014, 74, 2455–2464. [Google Scholar] [CrossRef] [Green Version]
- Naik, P.P.; Birbrair, A.; Bhutia, S.K. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell. Mol. Life Sci. 2019, 76, 27–43. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Nakhle, J.; Rodriguez, A.M.; Vignais, M.L. Multifaceted Roles of Mitochondrial Components and Metabolites in Metabolic Diseases and Cancer. Int. J. Mol. Sci. 2020, 21, 4405. [Google Scholar] [CrossRef]
- Macleod, K.F. Mitophagy and Mitochondrial Dysfunction in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 41–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [Green Version]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef] [Green Version]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef]
- Brillantes, M.; Beaulieu, A.M. Memory and Memory-Like NK Cell Responses to Microbial Pathogens. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Lim, S.A.; Kim, T.J.; Lee, J.E.; Sonn, C.H.; Kim, K.; Kim, J.; Choi, J.G.; Choi, I.K.; Yun, C.O.; Kim, J.H.; et al. Ex vivo expansion of highly cytotoxic human NK cells by cocultivation with irradiated tumor cells for adoptive immunotherapy. Cancer Res. 2013, 73, 2598–2607. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [Green Version]
- Kidger, A.M.; Rushworth, L.K.; Stellzig, J.; Davidson, J.; Bryant, C.J.; Bayley, C.; Caddye, E.; Rogers, T.; Keyse, S.M.; Caunt, C.J. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lee, D.A.; Wang, Y.; Wang, L.; Yao, Y.; Lin, Z.; Cheng, J.; Zhu, S. Membrane-bound interleukin-21 and CD137 ligand induce functional human natural killer cells from peripheral blood mononuclear cells through STAT-3 activation. Clin. Exp. Immunol. 2013, 172, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.M.; Vakili, M.R.; Molavi, O.; Lavasanifar, A. Self-Associating Poly(ethylene oxide)-block-poly(α-carboxyl-ε-caprolactone) Drug Conjugates for the Delivery of STAT3 Inhibitor JSI-124: Potential Application in Cancer Immunotherapy. Mol. Pharm. 2017, 14, 2570–2584. [Google Scholar] [CrossRef]
- Lee, K.M.; Forman, J.P.; McNerney, M.E.; Stepp, S.; Kuppireddi, S.; Guzior, D.; Latchman, Y.E.; Sayegh, M.H.; Yagita, H.; Park, C.K.; et al. Requirement of homotypic NK-cell interactions through 2B4(CD244)/CD48 in the generation of NK effector functions. Blood 2006, 107, 3181–3188. [Google Scholar] [CrossRef] [Green Version]
- Siewiera, J.; Gouilly, J.; Hocine, H.R.; Cartron, G.; Levy, C.; Al-Daccak, R.; Jabrane-Ferrat, N. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat. Commun. 2015, 6, 10183. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qian, P.; Jackson, F.R.; Qian, G.; Wu, G. Sequential activation of JAKs, STATs and xanthine dehydrogenase/oxidase by hypoxia in lung microvascular endothelial cells. Int. J. Biochem. Cell Biol. 2008, 40, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 411–433. [Google Scholar] [CrossRef]
- Deenick, E.K.; Pelham, S.J.; Kane, A.; Ma, C.S. Signal Transducer and Activator of Transcription 3 Control of Human T and B Cell Responses. Front. Immunol. 2018, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Matthews, J.; Parvin, S.; Sarosiek, K.A.; Zhao, D.; Jiang, X.; Isik, E.; Letai, A.; Lossos, I.S. Direct and immune-mediated cytotoxicity of interleukin-21 contributes to antitumor effects in mantle cell lymphoma. Blood 2015, 126, 1555–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dratwa, M.; Wysoczańska, B.; Łacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT—Regulation and Roles in Cancer Formation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Salmond, R.J. mTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Guan, D.; Wang, S.; Chai, L.Y.A.; Xu, S.; Lam, K.P. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front. Immunol. 2020, 11, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pol, J.G.; Caudana, P.; Paillet, J.; Piaggio, E.; Kroemer, G. Effects of interleukin-2 in immunostimulation and immunosuppression. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazina, T.; Shemesh, A.; Brusilovsky, M.; Porgador, A.; Campbell, K.S. Regulation of the Functions of Natural Cytotoxicity Receptors by Interactions with Diverse Ligands and Alterations in Splice Variant Expression. Front. Immunol. 2017, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Tejado, M.; Naranjo-Suarez, S.; Jimenez, C.; Carrera, A.C.; Landazuri, M.O.; del Peso, L. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: Protective role in apoptosis. J. Biol. Chem. 2001, 276, 22368–22374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Dolloff, N.G.; Dicker, D.T.; Malysz, J.; El-Deiry, W.S. CDK1 stabilizes HIF-1alpha via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle 2013, 12, 3689–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrien-Elliott, M.M.; Romee, R.; Fehniger, T.A. Improving natural killer cell cancer immunotherapy. Curr. Opin. Organ. Transpl. 2015, 20, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietra, G.; Vitale, C.; Pende, D.; Bertaina, A.; Moretta, F.; Falco, M.; Vacca, P.; Montaldo, E.; Cantoni, C.; Mingari, M.C.; et al. Human natural killer cells: News in the therapy of solid tumors and high-risk leukemias. Cancer Immunol. Immunother. 2016, 65, 465–476. [Google Scholar] [CrossRef]
- Kim, T.J.; Kim, N.; Kang, H.J.; Kim, E.O.; Kim, S.T.; Ahn, H.S.; Bluestone, J.A.; Lee, K.M. FK506 causes cellular and functional defects in human natural killer cells. J. Leukoc. Biol. 2010, 88, 1089–1097. [Google Scholar] [CrossRef]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.-J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.-W.; et al. Defective Localization With Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Yang, E.G.; Park, H. Hypoxia enhances the expression of prostate-specific antigen by modifying the quantity and catalytic activity of Jumonji C domain-containing histone demethylases. Carcinogenesis 2013, 34, 2706–2715. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.; Lee, S.; Park, B.; Park, H. Distinct hypoxic regulation of preadipocyte factor-1 (Pref-1) in preadipocytes and mature adipocytes. Biochim. Biophys. Acta 2018, 1865, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, S.; Ahn, B.Y.; Byun, K.; Cho, Y.M.; Yu, M.H.; Lee, B.; Hwang, D.; Park, K.S. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci. Signal. 2013, 6, rs4. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.A.; Moon, Y.; Shin, M.H.; Kim, T.-J.; Chae, S.; Yee, C.; Hwang, D.; Park, H.; Lee, K.-M. Hypoxia-Driven HIF-1α Activation Reprograms Pre-Activated NK Cells towards Highly Potent Effector Phenotypes via ERK/STAT3 Pathways. Cancers 2021, 13, 1904. https://doi.org/10.3390/cancers13081904
Lim SA, Moon Y, Shin MH, Kim T-J, Chae S, Yee C, Hwang D, Park H, Lee K-M. Hypoxia-Driven HIF-1α Activation Reprograms Pre-Activated NK Cells towards Highly Potent Effector Phenotypes via ERK/STAT3 Pathways. Cancers. 2021; 13(8):1904. https://doi.org/10.3390/cancers13081904
Chicago/Turabian StyleLim, Seon Ah, Yunwon Moon, Min Hwa Shin, Tae-Jin Kim, Sehyun Chae, Cassian Yee, Daehee Hwang, Hyunsung Park, and Kyung-Mi Lee. 2021. "Hypoxia-Driven HIF-1α Activation Reprograms Pre-Activated NK Cells towards Highly Potent Effector Phenotypes via ERK/STAT3 Pathways" Cancers 13, no. 8: 1904. https://doi.org/10.3390/cancers13081904