
Feasibility of Optical Genome Mapping in Cytogenetic Diagnostics of Hematological Neoplasms: A New Way to Look at DNA
 , ,
, ,  , , and
, , and 
Abstract
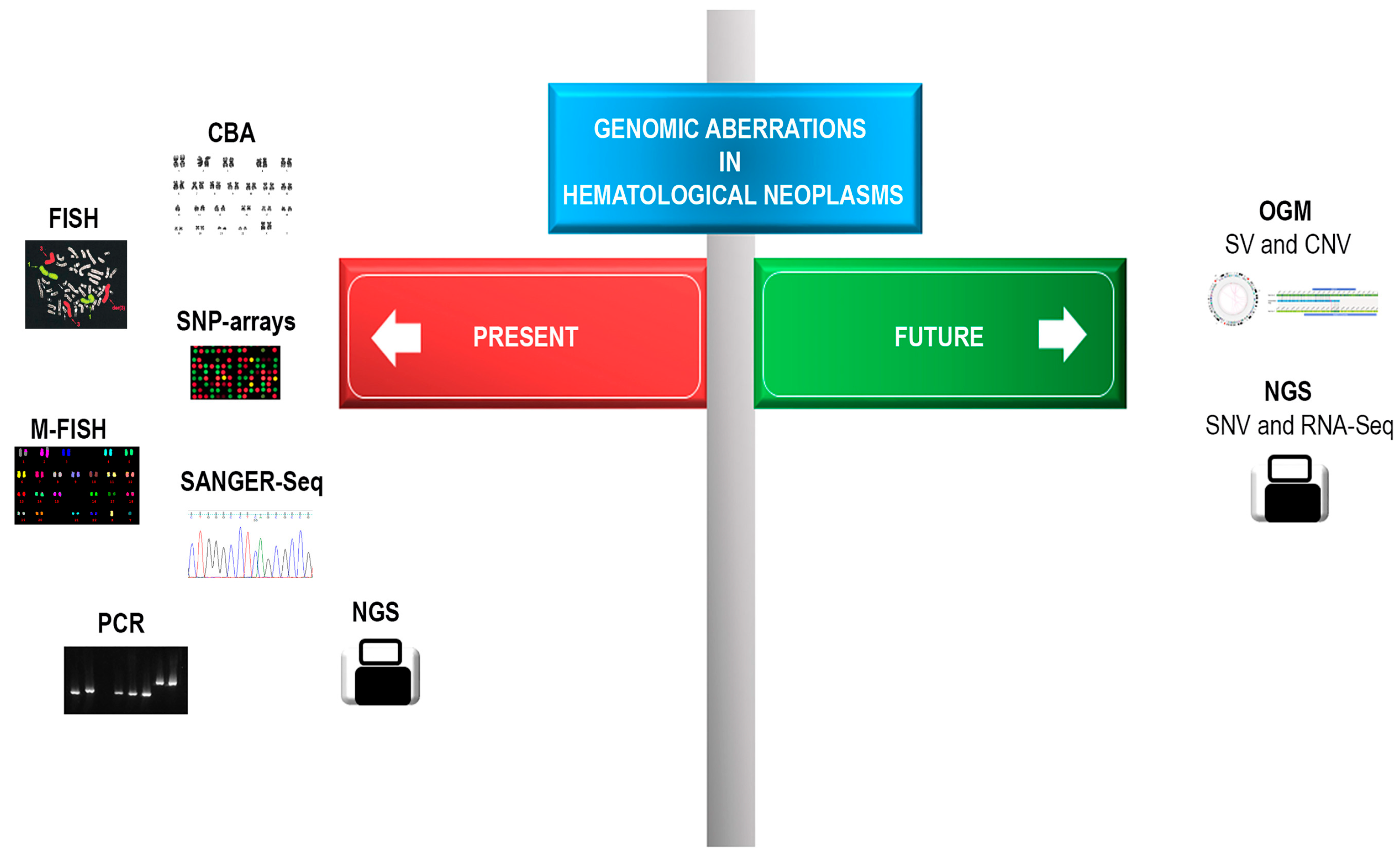
:1. Introduction
2. Advantages of OGM Technique
3. Limitations of OGM Technique
4. Application of OGM in Hematological Malignancies
4.1. Acute Myeloid Leukemia (AML) and Myelodysplastic Syndrome (MDS)
4.2. Acute Lymphoblastic Leukemia
4.3. Chronic Lymphocytic Leukemia
4.4. Multiple Myeloma
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical genome mapping in routine human genetic diagnostics—Its advantages and limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical genome mapping as a next-generation cytogenomic tool for detection of structural and copy number variations for prenatal genomic analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chung, C.Y.L.; Chan, T.F. Advances in optical mapping for genomic research. Comput. Struct. Biotechnol. J. 2020, 18, 2051–2062. [Google Scholar] [CrossRef]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural variation detection and analysis using bionano optical mapping. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1833, pp. 193–203. [Google Scholar]
- Dai, P.; Zhu, X.; Pei, Y.; Chen, P.; Li, J.; Gao, Z.; Liang, Y.; Kong, X. Evaluation of optical genome mapping for detecting chromosomal translocation in clinical cytogenetics. Mol. Genet. Genomic Med. 2022, 10, e1936. [Google Scholar] [CrossRef]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef]
- Bocklandt, S.; Hastie, A.; Cao, H. Bionano Genome Mapping: High-Throughput, Ultra-Long Molecule Genome Analysis System for Precision Genome Assembly and Haploid-Resolved Structural Variation Discovery. Adv. Exp. Med. Biol. 2019, 1129, 97–118. [Google Scholar]
- Dexter, T.M.; Allen, T.D.; Lajtha, L.G. Conditions controlling the proliferation of haemopoietic stem cells in vitro. J. Cell. Physiol. 1977, 91, 335–344. [Google Scholar] [CrossRef]
- Michaeli, J.; Lerer, I.; Rachmilewitz, E.A.; Fibach, E. Stimulation of proliferation of human myeloid leukemia cells in culture: Applications for cytogenetic analysis. Blood 1986, 68, 790–793. [Google Scholar] [CrossRef]
- Mrózek, K.; Harper, D.P.; Aplan, P.D. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol. Oncol. Clin. N. Am. 2009, 23, 991–1010. [Google Scholar] [CrossRef]
- Heerema, N.A.; Byrd, J.C.; Dal Cin, P.S.; Dell’ Aquila, M.L.; Koduru, P.R.K.; Aviram, A.; Smoley, S.A.; Rassenti, L.Z.; Greaves, A.W.; Brown, J.R.; et al. Stimulation of chronic lymphocytic leukemia cells with CpG oligodeoxynucleotide gives consistent karyotypic results among laboratories: A CLL Research Consortium (CRC) Study. Cancer Genet. Cytogenet. 2010, 203, 134–140. [Google Scholar] [CrossRef]
- Put, N.; Konings, P.; Rack, K.; Jamar, M.; Van Roy, N.; Libouton, J.M.; Vannuffel, P.; Sartenaer, D.; Ameye, G.; Speleman, F.; et al. Improved detection of chromosomal abnormalities in chronic lymphocytic leukemia by conventional cytogenetics using CpG oligonucleotide and interleukin-2 stimulation: A Belgian multicentric study. Genes Chromosomes Cancer 2009, 48, 843–853. [Google Scholar] [CrossRef]
- Mellors, P.W.; Binder, M.; Ketterling, R.P.; Greipp, P.T.; Baughn, L.B.; Peterson, J.F.; Jevremovic, D.; Pearce, K.E.; Buadi, F.K.; Lacy, M.Q.; et al. Metaphase cytogenetics and plasma cell proliferation index for risk stratification in newly diagnosed multiple myeloma. Blood Adv. 2020, 4, 2236–2244. [Google Scholar] [CrossRef]
- Dewald, G.W.; Kyle, R.A.; Hicks, G.A.; Greipp, P.R. The clinical significance of cytogenetic studies in 100 patients with multiple myeloma, plasma cell leukemia, or amyloidosis. Blood 1985, 66, 380–390. [Google Scholar] [CrossRef]
- Rovirosa, L.; Ramos-Morales, A.; Javierre, B.M. The Genome in a Three-Dimensional Context: Deciphering the Contribution of Noncoding Mutations at Enhancers to Blood Cancer. Front. Immunol. 2020, 11, 592087. [Google Scholar] [CrossRef]
- Montefiori, L.E.; Bendig, S.; Gu, Z.; Chen, X.; Pölönen, P.; Ma, X.; Murison, A.; Zeng, A.; Garcia-Prat, L.; Dickerson, K.; et al. Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov. 2021, 11, 2846–2867. [Google Scholar] [CrossRef]
- Rustad, E.H.; Yellapantula, V.D.; Glodzik, D.; Maclachlan, K.H.; Diamond, B.; Boyle, E.M.; Ashby, C.; Blaney, P.; Gundem, G.; Hultcrantz, M.; et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov. 2020, 1, 258–273. [Google Scholar] [CrossRef]
- Peterson, J.F.; Pitel, B.A.; Smoley, S.A.; Vasmatzis, G.; Smadbeck, J.B.; Greipp, P.T.; Ketterling, R.P.; Macon, W.R.; Baughn, L.B. Elucidating a false-negative MYC break-apart fluorescence in situ hybridization probe study by next-generation sequencing in a patient with high-grade B-cell lymphoma with IGH/MYC and IGH/BCL2 rearrangements. Cold Spring Harb. Mol. Case Stud. 2019, 5, a004077. [Google Scholar] [CrossRef]
- Smadbeck, J.; Peterson, J.F.; Pearce, K.E.; Pitel, B.A.; Figueroa, A.L.; Timm, M.; Jevremovic, A.D.; Shi, M.; Stewart, A.K.; Braggio, E.; et al. Mate pair sequencing outperforms fluorescence in situ hybridization in the genomic characterization of multiple myeloma. Blood Cancer J. 2019, 9, 103. [Google Scholar] [CrossRef]
- Lopes, J.L.; Webley, M.; Pitel, B.A.; Pearce, K.E.; Smadbeck, J.B.; Johnson, S.H.; Vasmatzis, G.; Sukov, W.R.; Greipp, P.T.; Hoppman, N.L.; et al. Characterizing false-positive fluorescence in situ hybridization results by mate-pair sequencing in a patient with chronic myeloid leukemia and progression to myeloid blast crisis. Cancer Genet. 2020, 243, 48–51. [Google Scholar] [CrossRef]
- Sharma, N.; Smadbeck, J.B.; Abdallah, N.; Zepeda-Mendoza, C.; Binder, M.; Pearce, K.E.; Asmann, Y.W.; Peterson, J.F.; Ketterling, R.P.; Greipp, P.T.; et al. The Prognostic Role of MYC Structural Variants Identified by NGS and FISH in Multiple Myeloma. Clin. Cancer Res. 2021, 27, 5430–5439. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Tiu, R.V.; Gondek, L.P.; O’Keefe, C.L.; Elson, P.; Huh, J.; Mohamedali, A.; Kulasekararaj, A.; Advani, A.S.; Paquette, R.; List, A.F.; et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood 2011, 117, 4552–4560. [Google Scholar] [CrossRef] [PubMed]
- Akkari, Y.M.N.; Bruyere, H.; Hagelstrom, R.T.; Kanagal-Shamanna, R.; Liu, J.; Luo, M.; Mikhail, F.M.; Pitel, B.A.; Raca, G.; Shago, M.; et al. Evidence-based review of genomic aberrations in B-lymphoblastic leukemia/lymphoma: Report from the cancer genomics consortium working group for lymphoblastic leukemia. Cancer Genet. 2020, 243, 52–72. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.; Wenger, G.D.; Chaubey, A.; Dash, D.P.; Kanagal-Shamanna, R.; Kantarci, S.; Kolhe, R.; Van Dyke, D.L.; Wang, L.; Wolff, D.J.; et al. Assessing copy number aberrations and copy-neutral loss-of-heterozygosity across the genome as best practice: An evidence-based review from the Cancer Genomics Consortium (CGC) working group for chronic lymphocytic leukemia. Cancer Genet. 2018, 228–229, 236–250. [Google Scholar] [CrossRef]
- Pugh, T.J.; Fink, J.M.; Lu, X.; Mathew, S.; Murata-Collins, J.; Willem, P.; Fang, M. Assessing genome-wide copy number aberrations and copy-neutral loss-of-heterozygosity as best practice: An evidence-based review from the Cancer Genomics Consortium working group for plasma cell disorders. Cancer Genet. 2018, 228–229, 184–196. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Hodge, J.C.; Tucker, T.; Shetty, S.; Yenamandra, A.; Dixon-McIver, A.; Bryke, C.; Huxley, E.; Lennon, P.A.; Raca, G.; et al. Assessing copy number aberrations and copy neutral loss of heterozygosity across the genome as best practice: An evidence based review of clinical utility from the cancer genomics consortium (CGC) working group for myelodysplastic syndrome, myelodysplastic/myeloproliferative and myeloproliferative neoplasms. Cancer Genet. 2018, 228–229, 197–217. [Google Scholar]
- Afable, M.G.; Wlodarski, M.; Makishima, H.; Shaik, M.; Sekeres, M.A.; Tiu, R.V.; Kalaycio, M.; O’Keefe, C.L.; Maciejewski, J.P. SNP array-based karyotyping: Differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood 2011, 117, 6876–6884. [Google Scholar] [CrossRef]
- Arenillas, L.; Mallo, M.; Ramos, F.; Guinta, K.; Barragán, E.; Lumbreras, E.; Larráyoz, M.J.; De Paz, R.; Tormo, M.; Abáigar, M.; et al. Single nucleotide polymorphism array karyotyping: A diagnostic and prognostic tool in myelodysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes Cancer 2013, 52, 1167–1177. [Google Scholar] [CrossRef]
- Meyer, C.; Larghero, P.; Almeida Lopes, B.; Burmeister, T.; Gröger, D.; Sutton, R.; Venn, N.C.; Cazzaniga, G.; Corral Abascal, L.; Tsaur, G.; et al. The KMT2A recombinome of acute leukemias in 2023. Leukemia 2023, 37, 988–1005. [Google Scholar] [CrossRef]
- Sanchez-Garcia, J.; Serrano, J.; Prados de La Torre, E.; Serrano-López, J.; Aparicio-Perez, C.; Barragán, E.; Montesinos, P. Identifying prognostic gene panels in acute myeloid leukemia. Expert Rev. Hematol. 2023, 16, 277–287. [Google Scholar] [CrossRef]
- Balducci, E.; Kaltenbach, S.; Villarese, P.; Duroyon, E.; Zalmai, L.; Friedrich, C.; Suarez, F.; Marcais, A.; Bouscary, D.; Decroocq, J.; et al. Optical genome mapping refines cytogenetic diagnostics, prognostic stratification and provides new molecular insights in adult MDS/AML patients. Blood Cancer J. 2022, 12, 126. [Google Scholar] [CrossRef]
- Pellestor, F.; Gaillard, J.B.; Schneider, A.; Puechberty, J.; Gatinois, V. Chromoanagenesis, the mechanisms of a genomic chaos. Semin. Cell Dev. Biol. 2022, 123, 90–99. [Google Scholar] [CrossRef]
- Shen, M.M. Chromoplexy: A New Category of Complex Rearrangements in the Cancer Genome. Cancer Cell 2013, 23, 567–569. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Ostapińska, K.; Styka, B.; Lejman, M. Insight into the Molecular Basis Underlying Chromothripsis. Int. J. Mol. Sci. 2022, 23, 3318. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Schroeder, M.C.; O’Laughlin, M.; Wilson, R.; MacMillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Hughes, A.E.O.; et al. Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. N. Engl. J. Med. 2021, 384, 924–935. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef]
- Rack, K.; De Bie, J.; Ameye, G.; Gielen, O.; Demeyer, S.; Cools, J.; De Keersmaecker, K.; Vermeesch, J.R.; Maertens, J.; Segers, H.; et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am. J. Hematol. 2022, 97, 548–561. [Google Scholar] [CrossRef]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The clinical utility of optical genome mapping for the assessment of genomic aberrations in acute lymphoblastic leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Lyons, E.; Town, C.D. Optical mapping in plant comparative genomics. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.; Lee, J.; Seo, J.; Park, C.K.; Shin, S.; Han, H.; Lee, S.T.; Choi, J.R.; Chung, B.H.; Choi, Y.D. Optical genome mapping identifies clinically relevant genomic rearrangements in prostate cancer biopsy sample. Cancer Cel. Int. 2022, 22, 306. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef]
- Podvin, B.; Roynard, P.; Boudry, A.; Guermouche, H.; Daudignon, A.; Terriou, L.; Bouabdelli, W.; Salameh, M.; Grardel, N.; Duployez, N.; et al. Whole-genome optical mapping to elucidate myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions. Leuk. Res. 2022, 123, 106972. [Google Scholar] [CrossRef]
- Wing Chun Pang, A.; Kosco, K.; Sahajpal, N.; Sridhar, A.; Hauenstein, J.; Clifford, B.; Estabrook, J.; Chitsazan, A.; Sahoo, T.; Iqbal, A.; et al. Clinical Validation of Optical Genome Mapping for the Detection of Structural Variations in Hematological Malignancies. medRxiv 2022. [Google Scholar] [CrossRef]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.M.N.; Chartrand, S.; LaBarge, B.; Claxton, D.F.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical Genome Mapping in Acute Myeloid Leukemia: A Multicenter Evaluation. Blood Adv. 2022, 7, 1297–13077. [Google Scholar] [CrossRef]
- Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; von Neuhoff, N.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. [Google Scholar] [CrossRef]
- Bueno, C.; Barrera, S.; Bataller, A.; Ortiz-Maldonado, V.; Elliot, N.; O’Byrne, S.; Wang, G.; Rovira, M.; Gutierrez-Agüera, F.; Trincado, J.L.; et al. CD34+CD19−CD22+ B-cell progenitors may underlie phenotypic escape in patients treated with CD19-directed therapies. Blood 2022, 140, 38–44. [Google Scholar] [CrossRef]
- Jean, J.; Kovach, A.E.; Doan, A.; Oberley, M.; Ji, J.; Schmidt, R.J.; Biegel, J.A.; Bhojwani, D.; Raca, G. Characterization of PAX5 intragenic tandem multiplication in pediatric B-lymphoblastic leukemia by optical genome mapping. Blood Adv. 2022, 6, 3343–3346. [Google Scholar] [CrossRef]
- Meyers, S.; Alberti-Servera, L.; Gielen, O.; Erard, M.; Swings, T.; De Bie, J.; Michaux, L.; Dewaele, B.; Boeckx, N.; Uyttebroeck, A.; et al. Monitoring of Leukemia Clones in B-cell Acute Lymphoblastic Leukemia at Diagnosis and during Treatment by Single-cell DNA Amplicon Sequencing. Hemasphere 2022, 6, E700. [Google Scholar] [CrossRef]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef]
- Ramos-Campoy, S.; Puiggros, A.; Kamaso, J.; Beà, S.; Bougeon, S.; Larráyoz, M.J.; Costa, D.; Parker, H.; Rigolin, G.M.; Blanco, M.L.; et al. TP53 Abnormalities Are Underlying the Poor Outcome Associated with Chromothripsis in Chronic Lymphocytic Leukemia Patients with Complex Karyotype. Cancers 2022, 14, 3715. [Google Scholar] [CrossRef]
- Gupta, A.; Place, M.; Goldstein, S.; Sarkar, D.; Zhou, S.; Potamousis, K.; Kim, J.; Flanagan, C.; Li, Y.; Newton, M.A.; et al. Single-molecule analysis reveals widespread structural variation in multiple myeloma. Proc. Natl. Acad. Sci. USA 2015, 112, 7689–7694. [Google Scholar] [CrossRef]
- Kriegova, E.; Fillerova, R.; Minarik, J.; Savara, J.; Manakova, J.; Petrackova, A.; Dihel, M.; Balcarkova, J.; Krhovska, P.; Pika, T.; et al. Whole-genome optical mapping of bone-marrow myeloma cells reveals association of extramedullary multiple myeloma with chromosome 1 abnormalities. Sci. Rep. 2021, 11, 14671. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Safavi, S.; Paulsson, K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: Two distinct subtypes with consistently poor prognosis. Blood 2017, 129, 420–423. [Google Scholar] [CrossRef]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef]
- Rigolin, G.M.; Cibien, F.; Martinelli, S.; Formigaro, L.; Rizzotto, L.; Tammiso, E.; Saccenti, E.; Bardi, A.; Cavazzini, F.; Ciccone, M.; et al. Chromosome aberrations detected by conventional karyotyping using novel mitogens in chronic lymphocytic leukemia with ‘normal’ FISH: Correlations with clinicobiologic parameters. Blood 2012, 119, 2310–2313. [Google Scholar] [CrossRef] [PubMed]
- González-Gascón-y-marín, I.; Muñoz-Novas, C.; Rodríguez-Vicente, A.E.; Quijada-álamo, M.; Hernández-Sánchez, M.; Pérez-Carretero, C.; Ramos-Ascanio, V.; Hernández-Rivas, J.Á. From biomarkers to models in the changing landscape of chronic lymphocytic leukemia: Evolve or become extinct. Cancers 2021, 13, 1782. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Matteo Rigolin, G.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef]
- Leeksma, A.C.; Baliakas, P.; Moysiadis, T.; Puiggros, A.; Plevova, K.; van der Kevie-Kersemaekers, A.M.; Posthuma, H.; Rodriguez-Vicente, A.E.; Tran, A.N.; Barbany, G.; et al. Genomic arrays identify high-risk chronic lymphocytic leukemia with genomic complexity: A multi-center study. Haematologica 2020, 105, 87–97. [Google Scholar] [CrossRef]
- Shah, G.L.; Landau, H.; Londono, D.; Devlin, S.M.; Kosuri, S.; Lesokhin, A.M.; Lendvai, N.; Hassoun, H.; Chung, D.J.; Koehne, G.; et al. Gain of chromosome 1q portends worse prognosis in multiple myeloma despite novel agent-based induction regimens and autologous transplantation. Leuk. Lymphoma 2017, 58, 1823–1831. [Google Scholar] [CrossRef]
- Aksenova, A.Y.; Zhuk, A.S.; Lada, A.G.; Zotova, I.V.; Stepchenkova, E.I.; Kostroma, I.I.; Gritsaev, S.V.; Pavlov, Y.I. Genome instability in multiple myeloma: Facts and factors. Cancers 2021, 13, 5949. [Google Scholar] [CrossRef]
- Hanamura, I. Multiple myeloma with high-risk cytogenetics and its treatment approach. Int. J. Hematol. 2022, 115, 762–777. [Google Scholar] [CrossRef]
- Maura, F.; Boyle, E.M.; Rustad, E.H.; Ashby, C.; Kaminetzky, D.; Bruno, B.; Braunstein, M.; Bauer, M.; Blaney, P.; Wang, Y.; et al. Chromothripsis as a pathogenic driver of multiple myeloma. Semin. Cell Dev. Biol. 2022, 123, 115–123. [Google Scholar] [CrossRef]
- Bhutani, M.; Foureau, D.M.; Atrash, S.; Voorhees, P.M.; Usmani, S.Z. Extramedullary multiple myeloma. Leukemia 2020, 34, 1–20. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M.; Dal Cin, P.; Diez Campelo, M.; Gallego, M.S.; Granada Font, I.; Haase, D.T.; et al. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2022, 139, 2273–2284. [Google Scholar] [CrossRef]
- Haferlach, T.; Schmidts, I. The power and potential of integrated diagnostics in acute myeloid leukaemia. Br. J. Haematol. 2020, 188, 36–48. [Google Scholar] [CrossRef]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; Casieri, P.; Rossi, A.R.; Vicari, L.; Liso, V.; Rocchi, M.; Specchia, G. Non random distribution of genomic features in breakpoint regions involved in chronic myeloid leukemia cases with variant t(9;22) or additional chromosomal rearrangements. Mol. Cancer 2010, 9, 120. [Google Scholar] [CrossRef]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef]
- De Haas, V.; Ismaila, N.; Advani, A.; Arber, D.A.; Dabney, R.S.; Patel-Donelly, D.; Kitlas, E.; Pieters, R.; Pui, C.H.; Sweet, K.; et al. Initial Diagnostic Work-Up of Acute Leukemia: ASCO Clinical Practice Guideline Endorsement of the College of American Pathologists and American Society of Hematology Guideline. J. Clin. Oncol. 2019, 37, 239–253. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627, quiz 3699. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| OGM Advantages | Limitations of Cytogenetic Standard Diagnostic Tests |
|---|---|
| Single assay instead of CBA, FISH, CMA, or MLPA | A cascade of several tests is required for a complete definition of cytogenetic alterations |
| Reduction in time and costs | CBA, FISH, CMA, and MLPA require more than 20 days and a few thousand dollars (USD) |
| High concordance with standard cytogenetic techniques | CBA often needs further confirmation |
| Detection of SVs (500 bp–1 Mbp) and CNVs (>0.5 Mb) at a VAF > 5–10% | CBA has a limit of about 5 Mb, FISH is based on target experiments, and CMA cannot reveal balanced alterations |
| Study of chromoanagenesis and better definition of complex karyotypes | Several cytogenetic tests are required to define complex rearrangements |
| OGM is not subject to culture bias | Both CBA and FISH require culture cells |
| Limitations of OGM | Cytogenetic Standard Diagnostic Test Advantages |
| Identification of false positive rearrangements | Some alterations identified via OGM are not confirmed via FISH or molecular analyses |
| False negative results | Conventional diagnostic tests are able to reveal Robertsonian translocations, telomere fusions, and isodicentric chromosomes |
| Inability to detect ploidy changes, CN-LOH, or single nucleotide variations (SNVs) | CBA and CMA are able to identify ploidy changes and CN-LOH |
| Failure to detect SVs down to a 5% allele fraction or located exclusively in centromeric/telomeric regions | CBA has the advantage of analyzing at the single-cell level and can provide information on clonal architecture |
| Hematological Malignancy | No. of Patients and Type of Cancer | OGM Application | OGM Limitations | Refs. |
|---|---|---|---|---|
| AML, MDS | 27 AML or MDS | Definition of complex rearrangements and identification of cryptic aberrations: NUP98::NSD1, MECOM::MSI2, and KMT2A::PTD | Failure to identify additional chromosome 21 because of sub-clonal occurrence | [49] |
| 27 MDS and 41 AML | Identification of recurrent complex rearrangements involving chromosomes 12 and 21 and novel cryptic cytogenetic abnormalities in AML: KMT2A::PTD, MYB alteration, and NUP98-rearrangement | Failure to identify some chromosomal alterations because of sub-clonal occurrence (Y loss and trisomy 8) or breakpoint localization in repetitive regions | [33] | |
| 100 AML | Identification of novel translocations: NSD1::NUP98 and ETV6::MECOM | Failure to detect sub-clonal anomalies | [50] | |
| 24 pediatric AML | Identification of new aberrations and new minimal residual disease markers | Failure to identify aneuploidies present in sub-clones and translocations occurring in repeated regions | [51] | |
| 101 MDS | Identification of several cryptic SVs in patients with normal karyotype and redefinition of risk-stratification | Failure to identity sub-clonal alterations | [39] | |
| ALL | 10 B-ALL or T-ALL | Identification of new fusions: LMO2::TCRA, TCRB::MYC, and IGH::CEBPB | Failure to identify four genomic anomalies because of poor coverage | [42] |
| 12 pediatric ALL | Better resolution of CNVs and identification of new rearrangements: t(9;11)(24.1;q22.1) and t(2;12;21)(p22.1;p13.2;q22.12) | Not reported | [41] | |
| 29 B-ALL and 12 T-ALL | Reclassification of three patients as B-ALL with TCF3::PBX1 and BCR::ABL1 like | Misinterpretation of two cases because of breakpoint localization in repetitive regions | [40] | |
| 10 CD19 CAR-treated B-ALL | Identification of genomic alterations on isolated CD34 + CD19-CD22+ cell population | Not reported | [52] | |
| 42 pediatric B-ALL | Characterization of PAX5-intagenic tandem multiplications | Not reported | [53] | |
| 12 B-ALL | Study of structural variants in combination with single-cell targeted DNA sequencing | Not reported | [54] | |
| CLL | 42 CLL | Definition of CLL genomic complexity and its use as a prognostic factor | Failure to identify 15–20% of genomic anomalies because of sub-clonal occurrence or breakpoint localization in repetitive regions | [55] |
| 9 CLL | Characterization of complex genomic alterations as chromothripsis and chromoplexy | Not reported | [56] | |
| MM | 1 MM | Integration between optical mapping and DNA genome sequencing to study MM progression | Use of a complex and time-consuming procedure for optical mapping | [57] |
| 4 EMM and 7 MM | Study of complex genomic architecture of CD138+ myeloma cells in newly diagnosed MM and EMM patients | Not reported | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coccaro, N.; Anelli, L.; Zagaria, A.; Tarantini, F.; Cumbo, C.; Tota, G.; Minervini, C.F.; Minervini, A.; Conserva, M.R.; Redavid, I.; et al. Feasibility of Optical Genome Mapping in Cytogenetic Diagnostics of Hematological Neoplasms: A New Way to Look at DNA. Diagnostics 2023, 13, 1841. https://doi.org/10.3390/diagnostics13111841
Coccaro N, Anelli L, Zagaria A, Tarantini F, Cumbo C, Tota G, Minervini CF, Minervini A, Conserva MR, Redavid I, et al. Feasibility of Optical Genome Mapping in Cytogenetic Diagnostics of Hematological Neoplasms: A New Way to Look at DNA. Diagnostics. 2023; 13(11):1841. https://doi.org/10.3390/diagnostics13111841
Chicago/Turabian StyleCoccaro, Nicoletta, Luisa Anelli, Antonella Zagaria, Francesco Tarantini, Cosimo Cumbo, Giuseppina Tota, Crescenzio Francesco Minervini, Angela Minervini, Maria Rosa Conserva, Immacolata Redavid, and et al. 2023. "Feasibility of Optical Genome Mapping in Cytogenetic Diagnostics of Hematological Neoplasms: A New Way to Look at DNA" Diagnostics 13, no. 11: 1841. https://doi.org/10.3390/diagnostics13111841