
Dendritic Cells as a Therapeutic Strategy in Acute Myeloid Leukemia: Vaccines
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Dendritic Cells: Ontogeny, Phenotypes, and Functions
3. Dendritic Cells Maturation and Antitumor Immunity Induction
4. Dendritic Cells-Based Therapies in Cancer: Vaccines
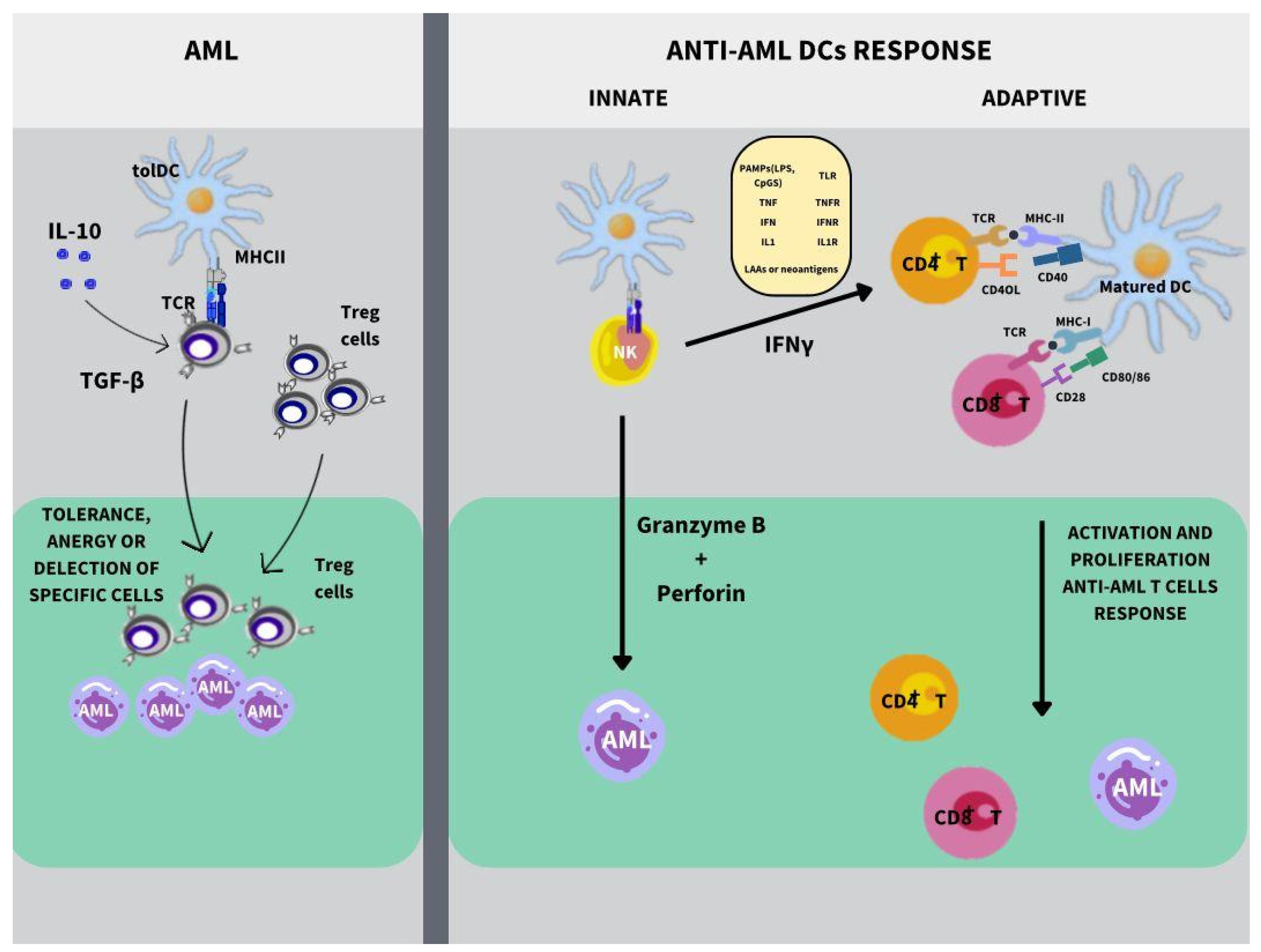
5. Dendritic Cells and Acute Myeloid Leukemia
5.1. Main Antigen Targets for Immunotherapy of AML
5.2. DC Vaccines in AML
5.2.1. Ex Vivo Loaded-DC Vaccines
5.2.2. In Vivo DC-Targeting Molecules Vaccine Carrier
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| AML-DCs | Dendritic cells derived from leukemic blasts |
| allo-HSCT | AllogeneicCD34+ hematopoietic stem cells trasplantation |
| APC | Antigen-presenting cells |
| cDCs | Conventional or myeloid dendritic cells |
| cDC1 | Conventional dendritic cells type 1 |
| cDC2 | Conventional dendritic cells type 2 |
| CLR | C-type lectin receptors |
| CML | Chronic myeloid leukemia |
| CMPs | Common myeloid progenitors |
| CR | Complete remission |
| CTAG1B | Cancer-testis antigen 1B |
| CTLA-4 | CytotoxicT lymphocytesAntigen 4 |
| CTLs | CytotoxicT lymphocytes |
| DAMPs | Danger-associated to molecular patterns |
| DCleu | Leukemia-derived dendritic cells |
| DCs | Dendritic cells |
| EVs | Tumor-derived exosomes |
| FDA | Food andDrugAdministration |
| FLT3-ITD | FMS-like tyrosine kinase 3 with internal tandem duplications |
| Flt3L | Fms related receptor tyrosine kinase 3 ligand |
| Gal-9 | Galectin-9 |
| GM-CSF | Granulocyte colony-stimulating factor- macrophages |
| HMAs | Hypomethylating agents |
| HSC | CD34+ hematopoietic stem cells |
| HSCT | CD34+ hematopoietic stem cells transplantation |
| IDH1 | IsocitrateDehydrogenase 1 |
| IDO | Indolamine 2,3-dioxygenase |
| IFN | Interferon |
| Ig | Immunoglobulins |
| IL | Interleukin |
| imDCs | Immature dendritic cells |
| IT | Immunotherapy |
| LAAs | Leukemia-associated antigens |
| LAMP | Lysosomal-associated membrane protein |
| LNP | Lipid nanoparticles |
| LRAs | Lineage-restricted antigens |
| LSA | Leukemia-specific antigens |
| MAGE | MelanomaAntigenGene |
| mDCs | Mature dendritic cells |
| MDS | Myelodysplastic syndrome |
| MDSCs | Immunosuppressive immature myeloid cells |
| MHC | Major histocompatibility complex |
| moDCs | Monocyte-derived dendritic cells |
| MPO | Myeloperoxidase |
| MRD | Minimal residual or measurable disease |
| MUC1 | Mucin1 |
| NK | Natural killer cells |
| NKT | Natural killerT cells |
| NPM1 | Nucleophosmin1 |
| NSE | Nonspecific esterase stains |
| NY-ESO-1 | TheCTANewYorkEsophagealSquamousCellCarcinoma-1 |
| PAMPs | Pathogen-associated molecular patterns |
| pDCs | Plasmacytoid dendritic cells |
| PD-L1 | ProgrammedDeath-ligand 1 |
| PGE | ProstaglandinE |
| PRAME | Preferentially expressedAntigen inMelanoma |
| ROS | Reactive oxygen species |
| shRNA | RNA interference fragment |
| SOCS1 | SuppressorOfCytokineSignaling 1 |
| SSB | Sudan blackB |
| STING | Stimulator of interferon genes complex |
| TCR | T cell receptors |
| Teff | EffectorT cells |
| TF | Transcription factors |
| TERT | Telomerase reverse transcriptase |
| Th | T helper cells |
| TLR | Toll like receptors |
| TNBC | Triple-negative breast cancer |
| Treg | RegulatoryT cells |
| VEGF | Vascular endothelial growth factor |
| VLPs | Virus-like particles |
| WT1 | WilmsTumor gene |
References
- Lee, J.M.; Lee, M.H.; Garon, E.; Goldman, J.W.; Salehi-Rad, R.; Baratelli, F.E.; Schaue, D.; Wang, G.; Rosen, F.; Yanagawa, J.; et al. Phase I Trial of Intratumoral Injection of CCL21 Gene-Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8(+) T-cell Infiltration. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4556–4568. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis e Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Audiger, C.; Chopin, M.; Nutt, S.L. Transcriptional regulation of dendritic cell development and function. Front. Immunol. 2023, 14, 1182553. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Versteven, M.; Lichtenegger, F.S.; Roex, G.; Campillo-Davo, D.; Lion, E.; Subklewe, M.; Van Tendeloo, V.F.; Berneman, Z.N.; Anguille, S. Dendritic Cell-Based Immunotherapy of Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 579. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Riether, C.; Schurch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, A.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Duan, L.J.; Wang, Q.; Zhang, C.; Yang, D.X.; Zhang, X.Y. Potentialities and Challenges of mRNA Vaccine in Cancer Immunotherapy. Front. Immunol. 2022, 13, 923647. [Google Scholar] [CrossRef]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lubbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- van der Hoorn, I.A.E.; Florez-Grau, G.; van den Heuvel, M.M.; de Vries, I.J.M.; Piet, B. Recent Advances and Future Perspective of DC-Based Therapy in NSCLC. Front. Immunol. 2021, 12, 704776. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Lee, Y.S.; O’Brien, L.J.; Walpole, C.M.; Pearson, F.E.; Leal-Rojas, I.M.; Masterman, K.A.; Atkinson, V.; Barbour, A.; Radford, K.J. Human CD141(+) dendritic cells (cDC1) are impaired in patients with advanced melanoma but can be targeted to enhance anti-PD-1 in a humanized mouse model. J. Immunother. Cancer 2021, 9, e001963. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3. [Google Scholar] [CrossRef]
- Barbullushi, K.; Rampi, N.; Serpenti, F.; Sciume, M.; Fabris, S.; De Roberto, P.; Fracchiolla, N.S. Vaccination Therapy for Acute Myeloid Leukemia: Where Do We Stand? Cancers 2022, 14, 2994. [Google Scholar] [CrossRef] [PubMed]
- Najafi, S.; Mortezaee, K. Advances in dendritic cell vaccination therapy of cancer. Biomed. Pharmacother. 2023, 164, 114954. [Google Scholar] [CrossRef]
- Strickland, S.A.; Vey, N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2022, 171, 103607. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallee, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e24. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed]
- Kvedaraite, E.; Ginhoux, F. Human dendritic cells in cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.F.; Alberti-Servera, L.; Eremin, A.; Grajales-Reyes, G.E.; Ivanek, R.; Tussiwand, R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat. Immunol. 2018, 19, 711–722. [Google Scholar] [CrossRef]
- Dress, R.J.; Dutertre, C.A.; Giladi, A.; Schlitzer, A.; Low, I.; Shadan, N.B.; Tay, A.; Lum, J.; Kairi, M.; Hwang, Y.Y.; et al. Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat. Immunol. 2019, 20, 852–864. [Google Scholar] [CrossRef]
- Solano-Galvez, S.G.; Tovar-Torres, S.M.; Tron-Gomez, M.S.; Weiser-Smeke, A.E.; Alvarez-Hernandez, D.A.; Franyuti-Kelly, G.A.; Tapia-Moreno, M.; Ibarra, A.; Gutierrez-Kobeh, L.; Vazquez-Lopez, R. Human Dendritic Cells: Ontogeny and Their Subsets in Health and Disease. Med. Sci. 2018, 6, 88. [Google Scholar] [CrossRef]
- Rodrigues, P.F.; Tussiwand, R. Novel concepts in plasmacytoid dendritic cell (pDC) development and differentiation. Mol. Immunol. 2020, 126, 25–30. [Google Scholar] [CrossRef]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B. Therapeutic potential of semi-mature dendritic cells for tolerance induction. Front. Immunol. 2012, 3, 123. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.U.; Khongorzul, P.; Raki, A.A.; Rajasekaran, A.; Gris, D.; Amrani, A. Dendritic Cells and Their Immunotherapeutic Potential for Treating Type 1 Diabetes. Int. J. Mol. Sci. 2022, 23, 4885. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Kautz, A.; Bohnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- Lauterbach, H.; Bathke, B.; Gilles, S.; Traidl-Hoffmann, C.; Luber, C.A.; Fejer, G.; Freudenberg, M.A.; Davey, G.M.; Vremec, D.; Kallies, A.; et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J. Exp. Med. 2010, 207, 2703–2717. [Google Scholar] [CrossRef]
- Hubert, M.; Gobbini, E.; Couillault, C.; Manh, T.V.; Doffin, A.C.; Berthet, J.; Rodriguez, C.; Ollion, V.; Kielbassa, J.; Sajous, C.; et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci. Immunol. 2020, 5, eaav3942. [Google Scholar] [CrossRef]
- Treilleux, I.; Blay, J.Y.; Bendriss-Vermare, N.; Ray-Coquard, I.; Bachelot, T.; Guastalla, J.P.; Bremond, A.; Goddard, S.; Pin, J.J.; Barthelemy-Dubois, C.; et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Gregorio, J.; Wang, Y.H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.J.; et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef]
- Oshi, M.; Newman, S.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Kalinski, P.; Endo, I.; Takabe, K. Plasmacytoid Dendritic Cell (pDC) Infiltration Correlate with Tumor Infiltrating Lymphocytes, Cancer Immunity, and Better Survival in Triple Negative Breast Cancer (TNBC) More Strongly than Conventional Dendritic Cell (cDC). Cancers 2020, 12, 3342. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Inaba, K.; Robbiani, D.F.; Steinman, R.M.; Muller, W.A. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity 1999, 11, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Marzaioli, V.; Canavan, M.; Floudas, A.; Wade, S.C.; Low, C.; Veale, D.J.; Fearon, U. Monocyte-Derived Dendritic Cell Differentiation in Inflammatory Arthritis Is Regulated by the JAK/STAT Axis via NADPH Oxidase Regulation. Front. Immunol. 2020, 11, 1406. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.V.; Lew, A.M.; Sutherland, R.M.; Zhan, Y. Monocyte-Derived Dendritic Cells Promote Th Polarization, whereas Conventional Dendritic Cells Promote Th Proliferation. J. Immunol. 2016, 196, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef]
- Wimmers, F.; Schreibelt, G.; Skold, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From in vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Yang, J.; Ronchese, F. Monocyte-Derived Dendritic Cells Are Essential for CD8(+) T Cell Activation and Antitumor Responses After Local Immunotherapy. Front. Immunol. 2015, 6, 584. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef]
- Sosa Cuevas, E.; Ouaguia, L.; Mouret, S.; Charles, J.; De Fraipont, F.; Manches, O.; Valladeau-Guilemond, J.; Bendriss-Vermare, N.; Chaperot, L.; Aspord, C. BDCA1(+) cDC2s, BDCA2(+) pDCs and BDCA3(+) cDC1s reveal distinct pathophysiologic features and impact on clinical outcomes in melanoma patients. Clin. Transl. Immunol. 2020, 9, e1190. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Woods, A.; Fu, T.; He, Q.; Nielsen, M.C.; Hasan, F.; Roszik, J.; Xiao, Z.; Vianden, C.; Khong, H.; et al. Cancer vaccine formulation dictates synergy with CTLA-4 and PD-L1 checkpoint blockade therapy. J. Clin. Investig. 2018, 128, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Schwepcke, C.; Klauer, L.K.; Deen, D.; Amberger, D.C.; Fischer, Z.; Doraneh-Gard, F.; Gunsilius, C.; Hirn-Lopez, A.; Kroell, T.; Tischer, J.; et al. Generation of Leukaemia-Derived Dendritic Cells (DC(leu)) to Improve Anti-Leukaemic Activity in AML: Selection of the Most Efficient Response Modifier Combinations. Int. J. Mol. Sci. 2022, 23, 8333. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, B.A.; Liang, J.C.; Thomas, E.K.; Flores-Romo, L.; Xie, Q.S.; Agusala, K.; Sutaria, S.; Sinha, I.; Champlin, R.E.; Claxton, D.F. Dendritic cells derived in vitro from acute myelogenous leukemia cells stimulate autologous, antileukemic T-cell responses. Blood 1999, 93, 780–786. [Google Scholar] [CrossRef]
- Dorrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic Cancer Vaccination with Ex Vivo RNA-Transfected Dendritic Cells-An Update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed]
- de Mey, W.; Locy, H.; De Ridder, K.; De Schrijver, P.; Autaers, D.; Lakdimi, A.; Esprit, A.; Franceschini, L.; Thielemans, K.; Verdonck, M.; et al. An mRNA mix redirects dendritic cells towards an antiviral program, inducing anticancer cytotoxic stem cell and central memory CD8(+) T cells. Front. Immunol. 2023, 14, 1111523. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Heiser, A.; Boczkowski, D.; Majumdar, A.; Naoe, M.; Lebkowski, J.S.; Vieweg, J.; Gilboa, E. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat. Med. 2000, 6, 1011–1017. [Google Scholar] [CrossRef]
- Khoury, H.J.; Collins, R.H., Jr.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D., 3rd; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Versteven, M.; Flumens, D.; Campillo-Davo, D.; De Reu, H.; Van Bruggen, L.; Peeters, S.; Van Tendeloo, V.; Berneman, Z.; Dolstra, H.; Anguille, S.; et al. Anti-Tumor Potency of Short-Term Interleukin-15 Dendritic Cells Is Potentiated by In Situ Silencing of Programmed-Death Ligands. Front. Immunol. 2022, 13, 734256. [Google Scholar] [CrossRef]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Johnson, D.B.; Puzanov, I.; Kelley, M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 2015, 7, 611–619. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Tureci, O.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Shi, J.; Huang, M.W.; Lu, Z.D.; Du, X.J.; Shen, S.; Xu, C.F.; Wang, J. Delivery of mRNA for regulating functions of immune cells. J. Control. Release Off. J. Control. Release Soc. 2022, 345, 494–511. [Google Scholar] [CrossRef]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med. J. 2020, 103, 38–40. [Google Scholar]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef]
- Stavrou, V.; Fultang, L.; Booth, S.; De Simone, D.; Bartnik, A.; Scarpa, U.; Gneo, L.; Panetti, S.; Potluri, S.; Almowaled, M.; et al. Invariant NKT cells metabolically adapt to the acute myeloid leukaemia environment. Cancer Immunol. Immunother. CII 2023, 72, 543–560. [Google Scholar] [CrossRef]
- Floisand, Y.; Remberger, M.; Bigalke, I.; Josefsen, D.; Valerhaugen, H.; Inderberg, E.M.; Olaussen, R.W.; Gjertsen, B.T.; Goedkoop, R.; Geiger, C.; et al. WT1 and PRAME RNA-loaded dendritic cell vaccine as maintenance therapy in de novo AML after intensive induction chemotherapy. Leukemia 2023, 37, 1842–1849. [Google Scholar] [CrossRef]
- O’Brien, L.J.; Guillerey, C.; Radford, K.J. Can Dendritic Cell Vaccination Prevent Leukemia Relapse? Cancers 2019, 11, 875. [Google Scholar] [CrossRef]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Abadir, E.; Gasiorowski, R.E.; Silveira, P.A.; Larsen, S.; Clark, G.J. Is Hematopoietic Stem Cell Transplantation Required to Unleash the Full Potential of Immunotherapy in Acute Myeloid Leukemia? J. Clin. Med. 2020, 9, 554. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Hassan, R.; Yaacob, N.S. Hypomethylating Agents and Immunotherapy: Therapeutic Synergism in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2021, 11, 624742. [Google Scholar] [CrossRef] [PubMed]
- Pearson, F.E.; Tullett, K.M.; Leal-Rojas, I.M.; Haigh, O.L.; Masterman, K.A.; Walpole, C.; Bridgeman, J.S.; McLaren, J.E.; Ladell, K.; Miners, K.; et al. Human CLEC9A antibodies deliver Wilms’ tumor 1 (WT1) antigen to CD141(+) dendritic cells to activate naive and memory WT1-specific CD8(+) T cells. Clin. Transl. Immunol. 2020, 9, e1141. [Google Scholar] [CrossRef] [PubMed]
- Sander, F.E.; Nilsson, M.; Rydstrom, A.; Aurelius, J.; Riise, R.E.; Movitz, C.; Bernson, E.; Kiffin, R.; Stahlberg, A.; Brune, M.; et al. Role of regulatory T cells in acute myeloid leukemia patients undergoing relapse-preventive immunotherapy. Cancer Immunol. Immunother. CII 2017, 66, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, M.; Sun, R.; Lyu, H.; Xiao, X.; Zhang, X.; Li, F.; Xie, D.; Xiong, X.; Wang, J.; et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, P.; Li, Z.; He, Y.; Gan, W.; Jiang, H. Anti-CLL1 Chimeric Antigen Receptor T-Cell Therapy in Children with Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3549–3555. [Google Scholar] [CrossRef]
- Saxena, K.; Herbrich, S.M.; Pemmaraju, N.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Pierce, S.A.; Jabbour, E.; Wang, S.A.; Bueso-Ramos, C.; et al. A phase 1b/2 study of azacitidine with PD-L1 antibody avelumab in relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 3761–3771. [Google Scholar] [CrossRef]
- Berdel, A.F.; Ruhnke, L.; Angenendt, L.; Wermke, M.; Rollig, C.; Mikesch, J.H.; Scheller, A.; Hemmerle, T.; Matasci, M.; Wethmar, K.; et al. Using stroma-anchoring cytokines to augment ADCC: A phase 1 trial of F16IL2 and BI 836858 for posttransplant AML relapse. Blood Adv. 2022, 6, 3684–3696. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Borthakur, G.; Pemmaraju, N.; Dinardo, C.D.; Kadia, T.M.; Jabbour, E.; Konopleva, M.; Macaron, W.; Ning, J.; Ma, J.; et al. A multi-arm phase Ib/II study designed for rapid, parallel evaluation of novel immunotherapy combinations in relapsed/refractory acute myeloid leukemia. Leuk. Lymphoma 2022, 63, 2161–2170. [Google Scholar] [CrossRef]
- Kreutmair, S.; Pfeifer, D.; Waterhouse, M.; Takacs, F.; Graessel, L.; Dohner, K.; Duyster, J.; Illert, A.L.; Frey, A.V.; Schmitt, M.; et al. First-in-human study of WT1 recombinant protein vaccination in elderly patients with AML in remission: A single-center experience. Cancer Immunol. Immunother. CII 2022, 71, 2913–2928. [Google Scholar] [CrossRef]
- Ho, V.T.; Kim, H.T.; Brock, J.; Galinsky, I.; Daley, H.; Reynolds, C.; Weber, A.; Pozdnyakova, O.; Severgnini, M.; Nikiforow, S.; et al. GM-CSF secreting leukemia cell vaccination for MDS/AML after allogeneic HSCT: A randomized, double-blinded, phase 2 trial. Blood Adv. 2022, 6, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Brayer, J.; Lancet, J.E.; Powers, J.; List, A.; Balducci, L.; Komrokji, R.; Pinilla-Ibarz, J. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am. J. Hematol. 2015, 90, 602–607. [Google Scholar] [CrossRef]
- Chevallier, P.; Saiagh, S.; Dehame, V.; Guillaume, T.; Peterlin, P.; Bercegeay, S.; Le Bris, Y.; Bossard, C.; Gauvrit, I.; Dreno, B.; et al. A phase I/II feasibility vaccine study by autologous leukemic apoptotic corpse-pulsed dendritic cells for elderly AML patients. Hum. Vaccines Immunother. 2021, 17, 3511–3514. [Google Scholar] [CrossRef]
- Wang, D.; Huang, X.F.; Hong, B.; Song, X.T.; Hu, L.; Jiang, M.; Zhang, B.; Ning, H.; Li, Y.; Xu, C.; et al. Efficacy of intracellular immune checkpoint-silenced DC vaccine. JCI Insight 2018, 3, e98368. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.; Van de Velde, A.L.; Nijs, G.; Braeckman, T.; Stein, B.; De Vries, J.M.; Berneman, Z.N.; Van Tendeloo, V.F. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 2009, 11, 653–668. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Rothe, M.; Deiser, K.; Altmann, T.; Bucklein, V.L.; Kohnke, T.; Augsberger, C.; Konstandin, N.P.; Spiekermann, K.; et al. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: Results of a phase I trial. Clin. Transl. Immunol. 2020, 9, e1117. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Srivastava, P.; Matsuzaki, J.; Brumberger, Z.; Wang, E.S.; Kocent, J.; Miller, A.; Roloff, G.W.; Wong, H.Y.; Paluch, B.E.; et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Amberger, D.C.; Schmetzer, H.M. Dendritic Cells of Leukemic Origin: Specialized Antigen-Presenting Cells as Potential Treatment Tools for Patients with Myeloid Leukemia. Transfus. Med. Hemother. 2020, 47, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Stone, R.M.; Uhl, L.; Neuberg, D.; Joyce, R.; Levine, J.D.; Arnason, J.; McMasters, M.; Luptakova, K.; Jain, S.; et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci. Transl. Med. 2016, 8, 368ra171. [Google Scholar] [CrossRef]
- Hernandez, S.S.; Jakobsen, M.R.; Bak, R.O. Plasmacytoid Dendritic Cells as a Novel Cell-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 11397. [Google Scholar] [CrossRef]
- van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. CII 2018, 67, 1505–1518. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar] [CrossRef]
- Wang, F.; Xiao, W.; Elbahnasawy, M.A.; Bao, X.; Zheng, Q.; Gong, L.; Zhou, Y.; Yang, S.; Fang, A.; Farag, M.M.S.; et al. Optimization of the Linker Length of Mannose-Cholesterol Conjugates for Enhanced mRNA Delivery to Dendritic Cells by Liposomes. Front. Pharmacol. 2018, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, S.; Duan, J.; Hu, Y.; Gu, N.; Xu, H.; Yang, X.D. Enhancement of DC-mediated anti-leukemic immunity in vitro by WT1 antigen and CpG co-encapsulated in PLGA microparticles. Protein Cell 2013, 4, 887–889. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Molecules | Delivery Methods | Achievements | References |
|---|---|---|---|
| hTERT | intradermal | hTERT-DCs ↑ survival among AML patients. | [60] |
| survivin-MUC1+flagelin | injection | SOCS1-silenced DCs ↑ safety and efficacy in treatment of relapsed AML. | [88] |
| WT1 mRNA | intradermal | Clinical response correlated with ↑ of WT1-specific CD8 T circulating cells. | [70,89] |
| WT1, PRAME, and CMVpp65 mRNA | intradermal | induction of leukaemia-specific primary and secundary immune responses. | [90] |
| leukemic apoptotic cells | intradermal | ↑ Median survival to double what was expected in AML patients. | [87] |
| WT1, PRAME mRNA | intradermal | induce antigen-specific T cell responses in AML patients. | [72] |
| CDX-1401 (anti-DEC205 and NY-ESO-1) | subcutaneous and intradermal | an antigen-specific immune response. | [91] |
| DCs Type | Phase | LAAs | CODE |
|---|---|---|---|
| Autologous TLR7/8-matured DCs | I | TLR7/8-matured DCs + mRNA-transfected encoding WT1, PRAME and CMVpp65 | NCT01734304 |
| Autologous DCs | I/II | Leukemic apoptotic corpse | NCT01146262 |
| Autologous DCs | I/II | mRNA-transfected encoding WT1 and PRAME | NCT02405338 |
| Autologous DCs/AML fusion cells | II | AML blasts | NCT01096602 |
| DCs +AML fusion cells | II | AML blasts | NCT03059485 |
| DCs | I | Overlapping peptides mixes derived from full-length NY-ESO-1, MAGE-A1, and MAGE-A3 | NCT01483274 |
| Autologous or HLA-matched donor-derived DCs | I | Expressing WT1/hTERT/Survivin | NCT05000801 |
| Autologous DCs | I | WT1 mRNA-transfected | NCT00834002 |
| Autologous DCs | II | GRNVAC1 transfected with mRNA encoding hTERT | NCT00510133 |
| Autologous DC + leukemic fusion cells | I | AML blasts | NCT00100971 |
| Autologous DCs | II | Electroporated with WT1 mRNA | NCT01686334 |
| Autologous DCs | I-II | Electroporated with WT1 mRNA | NCT03083054 |
| DCs + autologous leukemic cells | I | STING-Dependent Activators (STAVs) loaded autologous leukemic cells | NCT05321940 |
| Allogeneic DCs | II | DCP-001 | NCT03697707 |
| Allogeneic DCs | I-II | WT1 peptide-loaded | NCT00923910 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomares, F.; Pina, A.; Dakhaoui, H.; Leiva-Castro, C.; Munera-Rodriguez, A.M.; Cejudo-Guillen, M.; Granados, B.; Alba, G.; Santa-Maria, C.; Sobrino, F.; et al. Dendritic Cells as a Therapeutic Strategy in Acute Myeloid Leukemia: Vaccines. Vaccines 2024, 12, 165. https://doi.org/10.3390/vaccines12020165
Palomares F, Pina A, Dakhaoui H, Leiva-Castro C, Munera-Rodriguez AM, Cejudo-Guillen M, Granados B, Alba G, Santa-Maria C, Sobrino F, et al. Dendritic Cells as a Therapeutic Strategy in Acute Myeloid Leukemia: Vaccines. Vaccines. 2024; 12(2):165. https://doi.org/10.3390/vaccines12020165
Chicago/Turabian StylePalomares, Francisca, Alejandra Pina, Hala Dakhaoui, Camila Leiva-Castro, Ana M. Munera-Rodriguez, Marta Cejudo-Guillen, Beatriz Granados, Gonzalo Alba, Consuelo Santa-Maria, Francisco Sobrino, and et al. 2024. "Dendritic Cells as a Therapeutic Strategy in Acute Myeloid Leukemia: Vaccines" Vaccines 12, no. 2: 165. https://doi.org/10.3390/vaccines12020165