Haemolysis is decreased erythrocytes lifespan (normal about 100-120 days). The bone marrow responds to haemolysis by increasing erythrocyte production. Anaemia occurs only when the erythrocyte life span is reduced to about 15-20 days. A compensated haemolytic state is when a patient has a shorted erythrocyte lifespan that is adequately compensated by increased erythrocyte production. These patients may have features of haemolysis except anaemia.
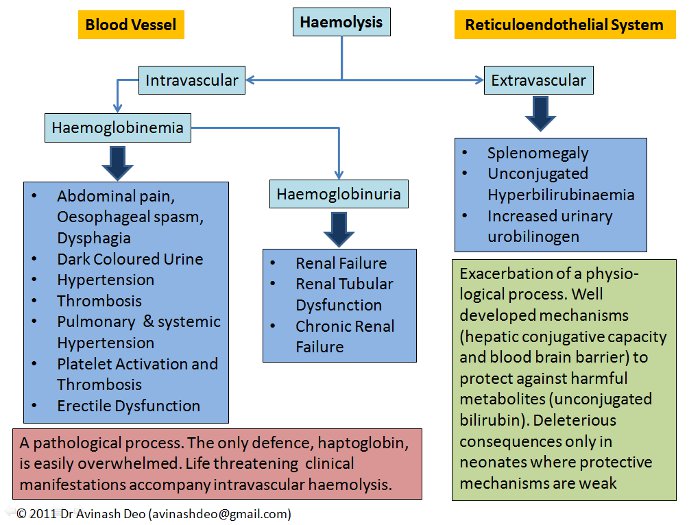
Figure 1. Manifestations of Haemolysis
Clinical Manifestations of Haemolysis
The site of haemolysis, intravascular or extravascular, determines the clinical manifestations of haemolysis (figure 1). Extravascular haemolysis is an exacerbation of a physiological catabolic process. The end product of extravascular haemolysis, like that of normal erythrocyte breakdown, is unconjugated bilirubin. Unconjugated bilirubin can cause brain damage in the forms of kernicterus. The conjugation of bilirubin is so efficient that the serum bilirubin rarely increase to more than 5mg/dL only due to bilirubin overproduction. The blood brain barrier prevents entry of bilirubin in the brain. Both the mechanisms are compromised in a neonate making them prone to kernicterus in case of haemolysis as is seen in haemolytic disease of the newborn. The only consequences of extravascular haemolysis occurring in patients beyond the neonatal period are anaemia, unconjugated hyperbilirubinaemia, mild to moderate splenomegaly and pigment gallstones.
Intravascular haemolysis is lysis of cells within the blood vessels. Cell free haemoglobin is oxidizing and pro-inflammatory. Haptoglobin binds and detoxifies cell free haemoglobin but it has a limited capacity. Haemoglobinaemia resulting from intravascular haemolysis causes serious clinical manifestations including renal failure, hypertension, smooth muscle spasm and a prothrombotic state. Many of these result from the nitric oxide scavenging capacity of haemoglobin (Rother et al JAMA 293:1653;2005). Persistent haemoglobinuria is a feature of chronic intravascular haemolysis, e.g. those with haemolysis due to prosthetic valves and paroxysmal nocturnal haemoglobinuria. Iron deficiency may accompany chronic intravascular haemolysis because of haemoglobinuria. Extravascular haemolysis is not associated with haemoglobin loss and does not cause iron deficiency.
Intravascular haemolysis often presents with acute severe haemolysis as may be seen with drug induced immune haemolysis, haemolysis in G6PD deficient patients, severe malaria and mismatched transfusion reactions. The diseases causing extravascular haemolysis have a less acute presentation.
The Diagnosis of Haemolytic Anaemia
Estimation of erythrocyte lifespan is too cumbersome to be used routinely in clinical practice. The diagnosis of haemolytic anaemia instead relies on indirect evidence of increased erythrocyte destruction. This includes:
- Increased production of erythrocytes in an anaemic patient with no evidence of blood loss
- Increase in products of erythrocyte destruction – Lactate dehydrogenase (LDH) and haemoglobin
- Consequences of haemoglobinaemia – low haptoglobin, haemoglobinuria, renal failure and hemosiderinuria
Increased erythrocyte production manifests as reticulocytosis (see The Reticulocyte Count for performing and interpreting reticulocyte count). If polycythaemia is not seen despite reticulocytosis, an equivalent erythrocyte loss is presumed to be occurring. Erythrocytes loss may internal, e.g., haemolysis, or external, e.g., bleeding. Reticulocytosis is seen when a large amount of blood is lost over a short period. Such losses are rarely occult. If there is no obvious blood loss, reticulocytosis in a patient whose haemoglobin is not increasing indicates haemolysis. Large occult haematomas, typically occurring retroperitoneally, may mimic haemolysis.
Lactate dehydrogenase (LDH) is abundant in brain, erythrocyte, liver and lung. Injury to any of these organs increases LDH. LDH is also increased in diseases characterized by increased cell proliferation e.g. acute leukaemia, lymphomas (particularly high grade non-Hodgkin lymphoma), myeloproliferative diseases and myelodysplastic syndrome. The clinical picture, radiology and laboratory investigations can exclude non-erythrocyte sources of LDH.
Haemolytic anaemias are characterized by an increase in LDH. The increase in LDH is more pronounced in patients with intravascular haemolysis and is matched by that seen in patients with nutritional megaloblastic anaemia. Unlike megaloblastic anaemia, intravascular haemolysis is characterized by reticulocytosis, haemoglobinaemia and haemoglobinuria.
The alterations caused by catabolism of haemoglobin released during haemolysis depend on the site of haemolysis (see haemoglobin catabolism). Haemoglobin released during extravascular haemolysis is metabolized to unconjugated bilirubin. The liver responds by increasing conjugation and excretion of bilirubin which increases urinary urobilinogen. Unconjugated hyperbilirubinaemia (unconjugated bilirubin >85% of total bilirubin) with increased urobilinogen is a characteristic of haemolytic anaemia. It differentiates haemolytic anaemia from Gilbert’s Syndrome, a common disorder of bilirubin conjugation that is seen in 3-7% of population. The urobilinogen levels in Gilbert’s syndrome are low. Crigler-Najjar syndrome type I and II are present with a more pronounced increase in bilirubin (>5mg/dL and >20mg/dL respectively) and are rarely considered as a differential diagnosis of an uncomplicated haemolytic anaemia. Bilirubin congugation defects do not show reticulocytosis. The bilirubin levels in patients with haemolytic anaemia rarely exceed 5mg/dL. Higher levels indicate a co-existing illness. If the bilirubin is unconjugated then an incidental co-inheritance of Gilbert’s syndrome should be suspected. Haemolytic anaemia increases the risk of pigment stones. These may cause obstructive jaundice. These patients, unlike those with haemolysis have a higher proportion of conjugated bilirubin with an elevated alkaline phosphatase. Donor screening for hepatitis B and C have made transfusion induced hepatitis a thing of the past.
Haemolysis releases haemoglobin. Cell free haemoglobin has oxidizing and pro-inflammatory properties. Haptoglobin is a haemoglobin scavenger that protects the body from the toxic effects of cell free haemoglobin. Haemolysis decreases serum haptoglobin (see haemoglobin catabolism). Extravascular haemolysis does not cause haemoglobinaemia, but contrary to what may be expected, it does cause low haptoglobin levels. Low haptoglobin is of no value in differentiating intravascular and extravascular haemolysis. It appears that there is some haemoglobinaemia during extravascular haemolysis. It is possible that there is some intravascular component to extravascular haemolysis or there is regurgitation of haemoglobin from the macrophage during phagocytosis. Haptoglobin is synthesized by the liver. Decrease synthesis limits the usefulness of haptoglobin in the diagnosis of haemolysis in the presence eof chronic liver disease. Haptoglobin is an acute phase reactant that in increased by corticosteroid administration. Patients with acute inflammation and those on corticosteroid therapy may have a normal haptoglobin despite haemolysis.
Haemoglobin filtered into the glomerular fluid results in haemoglobinuria when the absorptive capacity of renal tubules (5g/day, Turgeon, ML. Clinical Hematology: Theory and procedures 4th ed. Lippincott Williams and Wilkins, 2005:89) is exceeded. Haemoglobin can precipitate in the tubules and causes renal failure. The haemoglobin absorbed by the renal tubules in metabolized by haemoglobin oxygenase to iron, bilirubin and amino acids. The iron is stored in the renal tubular cells as hemosiderin. These cells desquamate and cause hemosiderinuria. Hemosiderinuria appears a few days after hemolysis and may persist for a week or more after hemolysis has abated. Iron deficiency anaemia caused by hemosiderinuria may complicate chronic low grade intravascular haemolysis as is seen in prosthetic valve haemolysis, paroxysmal nocturnal haemoglobinuria or march haemoglobinuria.
Diagnosis of haemolysis by transfusion requirements
Even with complete cessation of erythropoiesis only about one unit of blood is needed every week in an adult to maintain haemoglobin of about 10g/dL. A greater requirement in the absence of evidence of blood loss suggests haemolysis. Transfused blood will only be lysed if there is an extrinsic defect e.g. autoimmune haemolytic anaemia. Cells transfused to patients with an intrinsic defect e.g. hereditary spherocytosis will have a normal life span. A patient who needs more than one unit of blood per week to maintain haemoglobin of about 10g/dL is likely to have a haemolytic anaemia if blood loss can be excluded. This haemolysis is likely to be due to an extrinsic cause.
Differential Diagnosis of Haemolysis
Haemolysis may be confused with conditions associated with unconjugated hyperbilirubinaemia, anaemia and reticulocytosis. These include bilirubin conjugation defects, acute blood loss and megaloblastic anaemia.
Bilirubin Conjugation Defects Bilirubin conjugation defects include Gilbert’s syndrome and Crigler-Najjar syndrome types I and II. The bilirubin levels in Crigler-Najjar syndrome type I and II are more than 5mg/dL so these entities are rarely confused with haemolysis. Gilbert’s syndrome is characterized by bilirubin levels which can be seen in haemolysis but there is no associated reticulocytosis. Bilirubin conjugation defects are characterized by low urobilinogen levels.
Acute blood loss: Acute blood loss, like haemolysis, causes anaemia and reticulocytosis but these are not the presenting complains. Blood loss needed to cause reticulocytosis is rarely ignored by the patient and is in fact the reason for seeing the doctor. When severe, it may be accompanied by signs of hypovolemia. The presenting complains of patients with haemolytic anaemia are related to anaemia and hyperbilirubinaemia. Chronic occult blood loss has a different presentation that acute blood loss or haemolysis. The gradual fall in haemoglobin associated with occult blood loss makes the patients tolerant to anaemia unless a cardiovascular or respiratory co-morbidity exist. These patients present with a clinical picture of iron deficiency namely, fatigue, malaise and hypochromic microcytic anaemia with anisocytosis. There is no jaundice and there is reticulocytopenia. Large occult haematomas cause anaemia and reticulocytosis. As the haematoma resolves jaundice may be seen.
Megaloblastic anaemia Megaloblastic anaemia, like haemolytic anaemia, presents with macrocytic anaemia, indirect hyperbilirubinaemia, and a low serum haptoglobin. These patients have reticulocytopenia. The macrocytosis is more pronounced. An MCV of >110 fl usually indicates a megaloblastic anaemia. Patients with nutritional megaloblastic anaemia respond to treatment with pronounced reticulocytosis, usually seen by 5-7 days after initiating therapy and lasting for up to 2 weeks. Reticulocyte counts may be as high as 20-25%. A history of treatment with vitamin B12 and/or folic acid must be sought in every patient of haemolytic anaemia to avoid confusion with megaloblastic anaemia.
Related Pages
- Reticulocyte Count
- Evaluating Anaemia
- Haemoglobin Catabolism
- Sickle Haemoglobins and Variants
- Red Cell Indices
Haematology Atlas Pages

THANK YOU THAT WAS REALLY GOOD