
Abstract
Purpose of Review
Cardiac myosin inhibitors (CMIs) and activators are emerging therapies for hypertrophic cardiomyopathy (HCM) and heart failure with reduced ejection fraction (HFrEF), respectively. However, their effects on cardiac troponin levels, a biomarker of myocardial injury, are incompletely understood.
Recent Findings
In patients with HCM, CMIs cause substantial reductions in cardiac troponin levels which are reversible after stopping treatment. In patients with HFrEF, cardiac myosin activator (omecamtiv mecarbil) therapy cause modest increases in cardiac troponin levels which are reversible following treatment cessation and not associated with myocardial ischaemia or infarction.
Summary
Transient changes in cardiac troponin levels might reflect alterations in cardiac contractility and mechanical stress. Such transient changes might not indicate cardiac injury and do not appear to be associated with adverse outcomes in the short to intermediate term. Longitudinal changes in troponin levels vary depending on the population and treatment. Further research is needed to elucidate mechanisms underlying changes in troponin levels.
Similar content being viewed by others

Introduction
Cardiac myosin inhibitors and activators are emerging therapies developed for patients with hypertrophic cardiomyopathy (HCM) and heart failure with reduced ejection fraction (HFrEF), respectively. These treatments can affect cardiac troponin levels, a biomarker that is widely used to detect myocardial injury. The interpretation of troponin level changes during treatments can be challenging, as various factors can affect troponin levels. This article reviews the literature on differentiating cardiac troponin levels during cardiac myosin inhibition or activation treatments.
First, we discuss the biology of cardiac muscle which serves as the foundation for understanding cardiac myosin inhibitor and activator therapies. Second, we discuss the role of troponin assays, including in patients with HCM and HFrEF. Third, we describe cardiac myosin inhibitors in HCM and their effects on troponin levels. Fourth, we describe cardiac myosin activators in HFrEF and their effects on troponin levels. Fifth, we conclude by summarising guidance for interpreting troponin levels, and discuss future research directions.
Cardiac Muscle Biology
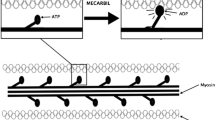
Striated muscles convert chemical energy to physical work and are comprised of skeletal muscle or cardiac muscle. A sarcomere is the basic contractile unit of cardiac muscle composed of thin actin and thick myosin myofilaments (Fig. 1). Muscle contraction occurs when myosin filaments pull actin filaments closer together, thus shortening the sarcomere unit.

Sarcomere, actin, myosin, and troponin
Cardiac myosin is the molecular motor that powers cardiac contraction by converting chemical energy from adenosine triphosphate (ATP) hydrolysis into mechanical force [1]. Cardiac myosin is a hexamer composed of two protein units of β- or α-myosin heavy chain and four myosin light chain molecules [2]. The heavy chains are responsible for ATP hydrolysis and force generation, while the light chains modulate myosin activity. The globular heads of myosin bind actin forming cross-bridges between the thick and thin filaments. The thin actin filament is closely associated with the regulatory troponin complex and α-tropomyosin. Cardiac myosin binding protein-C (MYBPC) contributes to the regulation of contraction.
Cardiac troponin is a protein complex of three subunits (troponin C; the Ca2+-binding subunit; troponin I, the inhibitory subunit; and troponin T, the tropomyosin-binding subunit) that regulate the contractility of cardiac muscle [3]. Troponin I and T are cardiac-specific isoforms that are released into the bloodstream after cardiac injury.
Troponin as a Biomarker
Over the last 2 decades, blood levels of cardiac troponin have transformed clinical care, as the standard biomarker to detect cardiac injury, most frequently used to diagnose or exclude acute coronary syndromes (ACS). Whilst troponin elevation is organ-specific for myocardial injury, it is not disease-specific and clinicians frequently encounter causes of troponin elevation other than ACS such as cardiomyopathies (e.g., HCM), heart failure (HF), tachycardia (e.g., atrial fibrillation), inflammation (e.g., myocarditis), renal dysfunction, anaemia, infection, and medications [4].
Cardiac troponin levels can be useful for diagnosis (ACS, myocarditis), screening (cardiac amyloidosis), prognosis (ACS, HF, HCM), monitor disease progression (myocarditis, HCM), and to monitor treatment efficacy and safety (coronary revascularisation, chemotherapy). For diagnosis, serial testing is recommended to rule out ACS. In patients with pericarditis, increased troponin levels could indicate myocardial involvement (myopericarditis). Elevated troponin levels might justify further evaluation for coronary artery disease, especially if not known prior, and if the pretest probability of coronary artery disease is higher. Elevated troponin levels may prompt clinicians to escalate or favour more intensive therapy during follow-up. A rise in troponin levels post-chemotherapy might indicate cardiotoxicity.
The universal definition of myocardial infarction, from its first version (2007) to its fourth version (2018), provides clinicians guidance on the interpretation of troponin elevations and serial changes (i.e., rise and fall) [Supplemental Table S1] [5•, 6•]. Troponin levels may remain elevated for days or weeks after the onset of cardiac injury, with the rate of declining troponin levels varying depending on disease severity, assay type, time interval measured, treatments received, and patient-specific clearance factors. It is important to consider patient- and laboratory-specific factors when interpretating cardiac troponin levels.
Patient factors such as age- and gender-specific reference ranges for troponin should be considered [7, 8]. Higher body mass index has been associated with increased likelihood of detectable cardiac troponin T (cTnT) levels [9]. A diurnal variation in cTnT has been described [10]. Cardiac troponin levels may be elevated postexercise even in apparently healthy individuals [11]. Troponin is attached to the myocyte contractile apparatus or detached from it in the cytosol—release of cytosolic troponin is proposed to account for the rise in troponin levels with exercise [12, 13].
Cardiac troponin assays vary between manufacturers and should not be used interchangeably. Using a single assay and a central core lab could help decrease variability, especially in multi-centre trials [6•]. A standardised approach is recommended to establish the 99th percentile upper reference limit (URL) for assays. False positive elevations in cardiac troponin can be caused by fibrin clots, heterophilic antibodies, alkaline phosphatase, rheumatoid factor, and cross-reactions of diagnostic (anti-cardiac troponin) antibodies with skeletal troponins [14]. Biotin supplementation may also interfere with troponin assays [15].
In the development of novel therapies, biomarkers such as cardiac troponin may help identify signals of myocardial damage earlier [16]. Avoiding myocardial injury or improving it could be a secondary clinical trial endpoint [17]. In cardio-oncology, elevated troponin pre-therapy is a risk factor for cancer therapy-related cardiomyopathy [18]. An increase in troponin after chemotherapy is a strong predictor of poor cardiac outcome. Cardiac troponin levels might help identify patients with a higher risk profile who are likely to benefit most from specific therapies. In a randomized study of 114 patients with a post-treatment (high-dose chemotherapy) troponin rise, treatment with enalapril seemed to prevent the development of late cardiotoxicity [19].
There are new developments in troponin assays. High sensitivity assays have become routinely available. Transdermal troponin has recently been shown to be clinically feasible for rapid, bloodless prediction of elevated cTnI levels in a study of 238 patients with ACS but clinical application remains a question and requires further investigation [20].
Interpreting Troponin Levels in HCM and HFrEF
HCM is a genetic heart muscle disease of the cardiac sarcomere. In approximately 40–50% of patients, HCM is caused by mutations in sarcomeric protein genes, most of which are in β-cardiac myosin (MYH7) and cardiac MYBPC [2]. HCM is characterised by myofilament disarray, myocardial hypercontractility, leading to left ventricular hypertrophy (unexplained by loading conditions), and fibrosis [21]. Proposed molecular mechanisms to explain the hypercontractile phenomenon include alterations in the actin-activated β-cardiac myosin chemo-mechanical ATPase cycle, an increased number of functionally accessible myosin heads (i.e., decrease in the super-relaxed state of myosin), and alterations in load dependence contractility that changes the power output of cardiac contraction [22, 23].
These pathophysiological changes contribute to left ventricular outflow tract (LVOT) obstruction, mitral regurgitation, diastolic dysfunction, myocardial ischaemia, arrhythmias, and autonomic dysfunction. These may cause exertional dyspnoea, fatigue, chest pain, exercise intolerance, palpitations, presyncope/syncope, and sudden cardiac death. Angina in the absence of epicardial coronary artery disease usually occurs with exertion and may result from inability of the coronary microcirculation to supply hypertrophied myocardium, and in obstructive HCM, high myocardial oxygen demand is associated with elevated left ventricular (LV) systolic pressure [24]. Adverse remodelling is defined by the presence of unfavourable structural modifications, translating into increasing LV fibrosis and worsening function, seen in about 15% to 20% of patients with HCM, a smaller proportion of whom will progress to HF [25].
Serum cardiac troponin is elevated in a significant proportion (ranging from 22 to 74%) of patients with HCM and is associated with clinical markers of disease severity including LVOT gradient (LVOT-G), left atrial diameter, LV mass, and fibrosis (as measured by late gadolinium enhancement on cardiovascular magnetic resonance imaging) [Supplemental Table S2]. In patients with HCM, elevated cTnT predicts clinical outcomes i.e., HF (hazard ratio 4.3 for New York Heart Association (NYHA) class II and hazard ratio 22.8 for NYHA class III), atrial fibrillation, and death [26]. Conversely, normal baseline cTnI has a 98% negative predictive value for adverse outcomes [26].
The clinical risk prediction model for sudden cardiac death in HCM (HCM Risk-SCD) recommended by the 2014 European Society of Cardiology (ESC) guidelines for HCM does not incorporate cardiac troponin [27]. However, the addition of cardiac troponin may be useful as an adjunct to current risk models in identifying patients with HCM and adverse cardiac remodelling [28].
To mitigate against postexercise elevations in cardiac troponin, clinical trial protocols generally have prespecified cardiac troponin testing prior to exercise stress echocardiography and avoiding significant activity prior to sample collection [29]. In a study of 127 patients with HCM versus 53 mutation carriers without hypertrophy (controls), patients with HCM were more likely to experience a postexercise increase in troponin compared with mutation carriers (18% vs 4%) [30]. In the HCM group, those who experienced a postexercise troponin increase had higher maximum heart rates and maximal wall thickness, and were more likely to have late gadolinium enhancement on cardiovascular magnetic resonance (CMR). Those with a postexercise increase in troponin were more likely to have high T2 measured on CMR. High T2 was the only independent predictor of troponin rise.
Many patients with HCM have been discouraged from exercising due to concerns about the risk of cardiac events. Several studies have examined the effect of exercise in patients with HCM. The Lifestyle and Exercise in HCM (LIVE-HCM) trial enrolled 1534 individuals with HCM and showed that those who exercised vigorously did not have an increased incidence of serious cardiac events over 3 years of follow-up compared with those who exercised moderately or were inactive [31, 32]. The Randomized Exploratory Study of Exercise Training in HCM (RESET-HCM) trial included 136 patients with HCM, and showed that moderate-intensity exercise compared with usual activity resulted in a small increase in exercise capacity as measured by peak oxygen consumption (pVO2) (between-group difference 1.27 [95% CI 0.17–2.37] mL/kg/min; p = 0.02) at 16 weeks [33].
Troponin levels are discussed in international guidelines for HCM and HF. In the 2014 ESC guidelines for HCM [34•], laboratory testing for troponin T is recommended as high levels of cTnT are associated with higher risk of cardiovascular events, HF, and death. The 2020 American College of Cardiology (ACC)/American Heart Association (AHA) guidelines for HCM do not provide any specific recommendations on the use of troponin levels [35•].
In the 2021 ESC guidelines for HF, initial laboratory exams recommended include troponin for exclusion of ACS, although elevated levels are detected in the vast majority of patients with AHF [36•]. In patients with suspected myocarditis, troponin is recommended as a mandatory diagnostic test because elevated troponins with dynamic changes are consistent with myocardial necrosis. In HF, persistently elevated troponin levels are a red flag for cardiac amyloidosis. In the 2022 AHA/ACC/Heart Failure Society of America (HFSA) guidelines for HF [18], evidence supporting stage B pre-HF includes patients with risk factors and persistently elevated cardiac troponin in the absence of competing diagnoses resulting in such biomarker elevations such as ACS, chronic kidney disease, pulmonary embolism, or myopericarditis.
Cardiac Myosin Inhibitors (CMIs) Development
HCM has become a treatable genetic heart disease with low mortality [37]. Traditionally, medical treatment for patients with HCM included beta-blockers, verapamil, diltiazem, and disopyramide as recommended in international guidelines [34•, 35•]. Treatment for HCM has been limited to symptomatic relief without tackling the root cause of the disease, excessive sarcomere contractility. Consequently, there is an unmet need for new therapies that can target the underlying pathophysiology of HCM.
CMIs have been recently developed as a therapy for HCM to directly reduce the myocardial hypercontractility that underlies the pathophysiology of HCM. CMIs target the myofilament apparatus to decrease the number of actin-myosin cross-bridges, thus resulting in dose-dependent reduction in contractility [38, 39]. As CMIs are a targeted disease-specific therapy, they promise less side-effects compared to non-targeted therapy [38, 40].
Mavacamten (formerly MYK-461) is the first-in-class, cardiac-specific, oral small molecule allosteric modulator of β-cardiac myosin that reversibly inhibits its binding to actin [41, 42]. Mavacamten has been approved by the U.S. Food and Drug Administration (FDA) in April 2022 to treat adults with NYHA class II–III obstructive HCM to improve exercise capacity and symptoms [43]. Aficamten (formerly CK-3773274 or CK-274) is a second in-class investigational oral small molecule allosteric inhibitor of cardiac myosin. The favourable pharmacokinetics of aficamten allows for rapid dose adjustments and rapid reversibility after discontinuation [Supplemental Table S3] [38, 41, 44]. Aficamten has minimal drug-drug interactions, with no significant cytochrome P inhibition or induction.
There are reported and ongoing trials investigating CMIs in patients with HCM summarized in Tables 1 and 2, and Fig. 2 and Supplemental Fig. 1A.

Percentage change from baseline in troponin levels in HCM randomized trials investigating cardiac myosin inhibitors (blue), HFrEF randomized trials investigating cardiac myosin activators (red), and observational studies of exercise (green). N indicates number of patients with data available at follow-up. If between-group difference is not reported: percentage change from baseline in troponin levels = ((change in intervention group—change in control group) / baseline value in intervention group) × 100%. In exercise studies, troponin levels were measured 24-h before and 3-h after 91-km mountain bike race. cTnI, cardiac troponin-I; cTnT, cardiac troponin-T; HCM, hypertrophic cardiomyopathy; HFrEF, heart failure with reduced ejection fraction; wks, weeks
Mavacamten completed trials
In a non-randomized clinical trial (A Phase 2 Open-label Pilot Study Evaluating MYK-461 in Subjects With Symptomatic HCM and LVOT Obstruction (PIONEER-HCM)) of 21 symptomatic patients with obstructive HCM, mavacamten reduced LVOT obstruction, improved exercise capacity, and symptoms [44]. Subsequently, a randomized, double-blind, placebo-controlled phase 3 trial, Mavacamten for Treatment of Symptomatic Obstructive HCM (EXPLORER-HCM), showed that mavacamten improved exercise capacity, symptoms, and LVOT obstruction in 251 patients with obstructive HCM [45••]. In the EXPLORER-HCM CMR substudy (35 patients), mavacamten treatment resulted in beneficial effects on cardiac remodelling, with reductions in absolute intracellular myocardial mass index, left ventricular mass index, maximum LV wall thickness, and left atrial volume indexed—all predictors of poor prognosis in obstructive HCM [46]. A Study to Evaluate Mavacamten in Adults with Symptomatic Obstructive HCM who are Eligible for Septal Reduction Therapy (VALOR-HCM) was a randomized double-blind, placebo-controlled phase 3 trial that enrolled 112 patients with obstructive HCM with intractable symptoms and showed that mavacamten significantly reduced the fraction of patients meeting guideline criteria for septal reduction therapy after 16 weeks [47••]. In a randomized double-blind, placebo-controlled, phase 2 study (Mavacamten in Adults With Symptomatic Non-Obstructive HCM (MAVERICK-HCM)) which randomized 59 subjects with symptomatic non-obstructive HCM with elevated N-terminal pro-B-type natriuretic peptide (NT-proBNP) ≥ 300 pg/mL, mavacamten was well tolerated and reduced NT-proBNP and cTnI levels but resulted in a reversible decline of LVEF ≤ 45% in 12.5% of patients [48••].
Mavacamten ongoing trials
The Extension Study of Mavacamten (MYK-461) in Adults With Symptomatic Obstructive HCM Previously Enrolled in PIONEER (PIONEER-OLE; NCT03496168) and A Long-Term Safety Extension Study of Mavacamten in Adults Who Have Completed MAVERICK-HCM or EXPLORER-HCM (MAVA-LTE; NCT03723655) studies are evaluating the long-term safety of mavacamten. The long-term extension study of VALOR-HCM is ongoing. A Study of Mavacamten in Non-Obstructive HCM (ODYSSEY-HCM; NCT05582395) is a randomized phase III trial designed to investigate the effect of mavacamten versus placebo on Kansas City Cardiomyopathy Questionnaire and pVO2.
Aficamten completed trials
In a phase I clinical trial in healthy adults, aficamten was well tolerated, adverse events were generally mild and comparable in frequency to those seen with placebo [39]. The double-blind, placebo-controlled, dose-finding Randomized Evaluation of Dosing With CK-274 in Obstructive Outflow Disease in HCM (REDWOOD-HCM) phase 2 trial enrolled a total of 95 patients across 4 cohorts. In cohorts 1 (5–15 mg) and 2 (10–30 mg) which enrolled 41 patients with obstructive HCM and LVOT obstruction, compared with placebo, aficamten reduced LVOT-G, paralleled by improvements in NT-proBNP at 10 weeks [49••]. In cohort 3 which recruited 13 patients with symptomatic obstructive HCM whose background therapy included disopyramide, aficamten treatment substantially reduced LVOT-G and improved NT-proBNP at 10 weeks [50••]. In cohort 4 which recruited 41 patients with non-obstructive HCM, aficamten treatment improved HF symptoms and NT-proBNP at 10 weeks [51••].
Aficamten ongoing trials
The Safety, Efficacy, and Quantitative Understanding of Obstruction Impact of Aficamten in HCM (SEQUOIA-HCM; NCT05186818) is an ongoing phase 3, randomized, placebo-controlled, double-blind trial assessing the efficacy and safety of aficamten on exercise capacity (pVO2 on cardiopulmonary exercise test), HF symptoms, and LVOT-G which has enrolled 282 symptomatic patients with obstructive HCM. The ongoing Follow-up, Open-Label, Research Evaluation of Sustained Treatment with Aficamten in HCM (FOREST-HCM; NCT04848506) trial is evaluating long-term outcomes with aficamten. A Phase 3, Multi-centre, Randomized, Double-blind Trial to Evaluate the Efficacy and Safety of Aficamten Compared to Metoprolol in Adults With Symptomatic Obstructive HCM (MAPLE-HCM; NCT05767346) is a trial with a head-to-head comparison of aficamten with the beta-blocker metoprolol succinate.
Clinical trials in HCM face several challenges. First, event rates of ‘hard endpoints’ e.g., mortality are relatively low, thus very large sample sizes or prolonged follow-up to accrue events would be required to power trials. Therefore, surrogate endpoints, such as how patients ‘feel and function’ endpoints, as well as biomarkers such as NT-proBNP and troponin levels, are used. Second, patients with HCM are heterogeneous and have a wide phenotype (obstructive versus non-obstructive; septal versus apical versus mid-ventricular hypertrophy; genotype positive versus negative; absence/presence of treatment with beta-blockers, calcium-channel blockers, and disopyramide, pre- versus post-septal reduction therapy). Third, CMIs reduce myocardial contractility, which may be clinically evident as a reduction in left ventricular ejection fraction (LVEF). Therefore, clinical trials of CMIs exclude patients with reduced LVEF, and incorporate rigorous echocardiographic monitoring for titration and safety, with down-titration and discontinuation criteria if reduced LVEF occurs. However, reduction in LVEF is in the mechanism of action of CMIs, and currently, it is unclear whether this excessive reversible reduction in LVEF is associated with adverse outcomes.
Effect of CMIs on Cardiac Troponin Levels
Several mavacamten trials have reported troponin outcomes (Tables 1 and 2) (Fig. 2 and Supplemental Fig. 1A). In EXPLORER-HCM, compared to placebo, mavacamten resulted in a significant and sustained reduction in cTnI levels over 30 weeks, even when LVOT-G did not decrease below commonly used thresholds to define LVOT obstruction [45••, 52]. In MAVERICK-HCM, compared to placebo, mavacamten resulted in rapid sustained improvements in cTnI concentrations over 16 weeks, particularly in those with elevated levels at baseline, despite no significant improvement in pVO2 or symptoms [48••]. In MAVERICK-HCM, cTnI results below the limit of detection (0.01 ng/mL) were imputed as one-half the limit (e.g., 0.005 ng/mL) for analysis which is one of the weaknesses of using troponin as an endpoint since many patients do not have elevated troponin level at baseline. Amongst those with elevated baseline cTnI in MAVERICK-HCM, after stopping study drug at week 16, cTnI levels increased to baseline levels by week 24. In the pooled mavacamten group in MAVERICK-HCM, change in NT-proBNP at week 4 correlated with change in cTnI at week 16 (r = 0.45, p = 0.006). In VALOR-HCM, compared to placebo, mavacamten reduced cTnI levels (geometric mean ratio difference 0.53 (95% CI 0.41 to 0.70), p < 0.001) [47••].
One aficamten trial (REDWOOD-HCM) with 4 cohorts has reported troponin levels. In cohorts 1 and 2, aficamten treatment was associated with marked reductions in NT-proBNP, and non-significant reductions in troponin levels (18% relative reduction (p = 0.29 compared with placebo) and 26% relative reduction (p = 0.097 compared with placebo) in cohorts 1 and 2, respectively). These findings suggest that aficamten may result in other potential downstream pathophysiologic benefits including decreases in LV wall stress and reduction in myocardial injury [49••]. Whether the mechanism of biomarker improvement is primarily related to normalization of LV systolic pressure, improved microvascular blood flow, or other downstream effects of direct myosin modulation requires further study [49••]. In cohort 3, patients on aficamten treatment trended to lower cTnI [50••]. In cohort 4, aficamten treatment reduced cTnI at each study visit up to 10 weeks, and after the 2-week washout period, cTnI levels returned to baseline levels [51••]. Interim analyses from FOREST-HCM also show reductions in cardiac troponin levels at 12 and 24 weeks [51••].
One hypothesis is that in patients with HCM, hypercontractility can result in elevated troponin levels in some patients, and treating the hypercontractility with CMIs can decrease troponin levels. Interpretation of troponin levels in patients with HCM on CMIs is dependent on the clinical presentation. CMIs are expected to lower troponin levels, but patients also typically become more active with improved symptoms on CMIs, where activity and exercise can lead to mild increases in troponin levels as well. Furthermore, HCM and CMIs should not result in a sharp and large increase in troponin levels, so other aetiologies should be investigated. Finally, incorporating LVEF with the troponin level ensures that one does not miss systolic dysfunction on CMIs as part of this troponin level fluctuation.
Cardiac Myosin Activator Development
Inotropic agents can be classified into cardiac calcitropes (which alter intracellular calcium concentrations), myotropes (which affect the molecular motor and scaffolding), and mitotropes (which influence energetics) [53]. Traditional inotropes (i.e., calcitropes) stimulate contractility via energetically costly augmentation of calcium cycling [54]. Inotropic drugs that acutely improve contractility and cardiac output have had unintended adverse outcomes with increased risk of myocardial ischaemia, ventricular arrhythmias, or death in trials [55,56,57]. These adverse outcomes are believed to be due to excessively increased cardiac energetic demand due to increased cyclic adenosine monophosphate (AMP) signalling and calcium cycling [58], in line with the hypothesis that the failing heart is energy starved [59].
Newer agents such as myotropes—small-molecule cardiac sarcomere activators that directly increase contractility—directly activate the sarcomere, independent of calcium i.e., without activating cyclic AMP signalling or increasing intracellular calcium cycling [53, 60]. Omecamtiv mecarbil (OM) and danicamtiv are selective cardiac myosin activators, also known as a cardiac myotropes, developed for the potential treatment of patients with HFrEF. As a myotrope, OM has no effect on calcium transients [60]. Cardiac myosin activators represent a novel therapeutic option for HFrEF.
Several trials have assessed OM and outcomes in patients with acute and chronic HFrEF which are summarized in Tables 3 and 4, and Fig. 2 and Supplemental Fig. 1B.
Acute HF trials
In the Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure (ATOMIC-AHF) trial enrolling 606 patients admitted for acute HF with LVEF ≤ 40%, a 48-h intravenous infusion of OM treatment did not improve dyspnoea but was generally well tolerated and increased systolic ejection time [61••].
Chronic HFrEF trials
In a phase 2 trial (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF)) enrolling 448 patients with stable, symptomatic chronic HF and LVEF ≤ 40%, oral OM dosing guided by pharmacokinetics achieved plasma concentrations, improved cardiac function, and decreased ventricular diameter over 20 weeks [62••]. The phase 3 trial (Global Approach to Lowering Adverse Cardiac Outcomes Through Improving Contractility in Heart Failure (GALACTIC-HF)) enrolled 8256 patients with chronic HF and LVEF ≤ 35% and showed that compared to placebo, those who received OM had a lower incidence of a composite of a HF event or cardiovascular death, over a median follow-up of 21.8 months [63••]. In a phase 3 trial (Effect of Omecamtiv Mecarbil on Exercise Capacity in Chronic Heart Failure With Reduced Ejection Fraction (METEORIC-HF)) including 276 patients with chronic HF and LVEF ≤ 35%, compared to placebo, OM did not significantly improve exercise capacity over 20 weeks [64]. In a phase 2a trial enrolling 40 patients with stable HFrEF, danicamtiv was well tolerated and improved LV systolic function and left atrial volume and function [12].
The U.S. FDA issued a briefing document on OM on December 13, 2022, noting that treatment with OM caused a small increase in cardiac biomarkers including cTnI and creatine kinase-MB but acknowledged that the clinical significance of these findings were unclear [65, 66]. However, despite the clear safety of OM in the previously mentioned trials, the small increase in cTnI was debated in the advisory committee meeting as a potential signal of harm. The U.S. FDA issued a complete response letter on February 28, 2023, communicating that GALACTIC-HF alone does not establish substantial evidence of effectiveness sufficient for approval of OM [67].
Effect of Cardiac Myosin Activators on Cardiac Troponin Levels
Several randomized trials have reported small increases in cardiac troponin levels following treatment with OM (Tables 3 and 4) (Fig. 2 and Supplemental Fig. 1B).
In ATOMIC-AHF, at well tolerated doses (< 1200 ng/mL) of OM, small increases in cTnI concentrations were noted in OM-treated patients compared with placebo (median difference at 48 h, 0.004 ng/mL) in the absence of other clinical evidence of myocardial ischaemia [61••]. However, there was no obvious relationship with OM concentration (p = 0.95).
In COSMIC-HF, around a quarter of enrolled patients had cTnI concentrations greater than the 99th percentile URL (0.04 ng/mL) at baseline, with proportions being similar across groups. At week 20, in patients receiving fixed-dose and titrated OM, there was a small increased concentration of circulating cTnI (0.001 ng/mL and 0.006 ng/mL, respectively, compared with no change seen in the placebo group) that did not correlate with the maximum plasma concentration of OM (r2 = 0.017) [62••]. Possible cardiac ischaemia or infarction were adjudicated by the study’s clinical events committee if investigators reported events suggestive of myocardial ischaemia or if cTnI concentration was > 99th percentile URL of 0·04 ng/mL when the previous concentration had been undetectable, or if the value had increased by > 0·03 ng/mL. Of 278 possible cardiac ischaemia or infarction events associated with increased cTnI concentrations, none of these were deemed to be myocardial infarction following adjudication by the clinical events committee [62••]. Increases in cTnI concentrations returned to baseline values after treatment was stopped.
In GALACTIC-HF, the median cTnI level was higher by 4 and 2 ng/L in the OM group compared to the placebo group, at weeks 24 and 48, respectively [63••]. The incidences of myocardial ischaemia, ventricular arrhythmias, and death were similar in both groups with almost 7500 patient-years of follow-up. Furthermore, no detrimental effects of OM were detected with respect to blood pressure, heart rate, creatinine, or potassium levels.
Although these trials found a small increase in plasma levels of troponin, treatment with OM did not increase the risk of clinical adverse events. The magnitude of troponin release is small in comparison to troponin release in response to exercise in healthy endurance athletes [11] and within the limits of diurnal variation for patients without HF [68]. Excessive exposure to OM may result in prolongation of the systolic ejection time to an extent that theoretically would reduce diastolic coronary blood flow, thus precipitating myocardial ischaemia or infarction [64]. None of the increases in cTnI concentration in the OM program were deemed to indicate myocardial ischaemia, and occurred in the context of improving systolic function, decreasing ventricular volumes, and declining NT-proBNP concentrations. The more likely hypothesis is that further sarcomere recruitment and activation by OM results in the very small increase in troponin observed in OM trials. In comparison, exercise in athletes results in a much higher magnitude in troponin release, although more acute and repetitive rather than chronic elevation, as shown in Fig. 2. Whether other mechanisms are involved, such as exosomal trafficking, requires further investigation [69].
Conclusion
In patients with both obstructive and non-obstructive HCM, CMIs reduce cardiac troponin levels over short- to medium-term follow-up (10–30 weeks). These reductions in troponin levels are consistent, profound, reversible (after stopping treatment and washout period), and associated with improvements in symptoms, functional capacity, NT-proBNP, and LVOT-G. Long-term data will be critical for characterising the durability of benefit and safety of CMIs to inform potential lifelong therapy in patients with HCM.
In patients with HFrEF, the cardiac myosin activator OM causes a small rise in cardiac troponin levels, which was seen both with acute and chronic treatment, and reversible following treatment cessation. However, these small increases in troponin levels were not associated with increased risk of adjudicated clinical myocardial ischaemia, ventricular arrhythmias, or death, providing reassurance that the biochemical changes do not necessarily equate to adverse clinical outcomes. In February 2023, the U.S. FDA declined the approval of OM for patients with chronic HFrEF [67]. This decision was perhaps, at least in part, swayed by concerns relating to the small and reversible increases in cardiac troponin levels following OM therapy, despite benefits seen in reduction of HF events and cardiovascular death and no evidence of increase in clinical adverse events. This is an example where the use of a biomarker like troponin, if taken out of context, can be detrimental. Exercise leads to significant increase in troponin in the setting of robust sarcomere recruitment that far exceeds what is seen with OM, and appropriately we do not claim that exercise is potentially dangerous due to this rise in troponin.
Troponin levels can be helpful to guide clinicians prior to, during, and after starting cardiac myosin modulators. The predictable rise and fall in cardiac troponin levels might provide an indication of therapeutic efficacy, safety, and importantly, patient adherence with therapy. Troponin levels could also be considered in the future as means to sub-select a population with substantial disease burden. The data accrued in this area are vital given the incredibly high utilization of troponin assays (appropriately and inappropriately), coupled with a likely exponential use of these novel cardiac myosin modulators in routine clinical care.
Data Availability
All supporting data are available within the cited references. No new data were generated in support of this review.
Change history
01 December 2023
A Correction to this paper has been published: https://doi.org/10.1007/s11897-023-00639-5
Abbreviations
- ACC:
-
American College of Cardiology
- ACS:
-
Acute coronary syndrome
- AHA:
-
American Heart Association
- AMP:
-
Adenosine monophosphate
- ATOMIC-AHF:
-
Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure
- ATP:
-
Adenosine triphosphate
- CMI:
-
Cardiac myosin inhibitor
- CMR:
-
Cardiovascular magnetic resonance
- COSMIC-HF:
-
Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure
- cTnI:
-
Cardiac troponin-I
- cTnT:
-
Cardiac troponin-T
- EMBARK-HFpEF:
-
A Study of Mavacamten in Participants With Heart Failure With Preserved Ejection Fraction and Elevation of NT-proBNP With or Without Elevation of cTnT
- ESC:
-
European Society of Cardiology
- EXPLORER-HCM:
-
Mavacamten for Treatment of Symptomatic Obstructive HCM
- FDA:
-
Food and Drug Administration
- FOREST-HCM:
-
Follow-up, Open-Label, Research Evaluation of Sustained Treatment with Aficamten in HCM
- GALACTIC-HF:
-
Global Approach to Lowering Adverse Cardiac Outcomes Through Improving Contractility in Heart Failure
- HCM:
-
Hypertrophic cardiomyopathy
- HF:
-
Heart failure
- HFrEF:
-
Heart failure with reduced ejection fraction
- HFSA:
-
Heart Failure Society of America
- ICD:
-
Implantable cardioverter defibrillator
- KCCQ:
-
Kansas City Cardiomyopathy Questionnaire
- LIVE-HCM:
-
Lifestyle and Exercise in HCM
- LV:
-
Left ventricular
- LVEF:
-
Left ventricular ejection fraction
- LVOT:
-
Left ventricular outflow tract
- LVOT-G:
-
Left ventricular outflow tract gradient
- MAPLE-HCM:
-
A Phase 3, Multi-centre, Randomized, Double-blind Trial to Evaluate the Efficacy and Safety of Aficamten Compared to Metoprolol in Adults With Symptomatic Obstructive HCM
- MAVA-LTE:
-
A Long-Term Safety Extension Study of Mavacamten in Adults Who Have Completed MAVERICK-HCM or EXPLORER-HCM
- MAVERICK-HCM:
-
Mavacamten in Adults With Symptomatic Non-Obstructive HCM
- METEORIC-HF:
-
Effect of Omecamtiv Mecarbil on Exercise Capacity in Chronic Heart Failure With Reduced Ejection Fraction
- MYBPC:
-
Myosin-binding protein C
- NT-proBNP:
-
N-terminal pro-B-type natriuretic peptide
- NYHA:
-
New York Heart Association
- ODYSSEY-HCM:
-
A Study of Mavacamten in Non-Obstructive HCM
- OM:
-
Omecamtiv mecarbil
- PIONEER-HCM:
-
A Phase 2 Open-label Pilot Study Evaluating MYK-461 in Subjects With Symptomatic HCM and Left Ventricular Outflow Tract Obstruction
- PIONEER-OLE:
-
Extension Study of Mavacamten (MYK-461) in Adults With Symptomatic Obstructive HCM Previously Enrolled in PIONEER
- pVO2 :
-
Peak oxygen consumption
- REDWOOD-HCM:
-
Randomized Evaluation of Dosing With CK-274 in Obstructive Outflow Disease in HCM
- RESET-HCM:
-
Randomized Exploratory Study of Exercise Training in HCM
- SEQUOIA-HCM:
-
Safety, Efficacy, and Quantitative Understanding of Obstruction Impact of Aficamten in HCM
- URL:
-
Upper reference limit
- VALOR-HCM:
-
A Study to Evaluate Mavacamten in Adults with Symptomatic Obstructive HCM who are Eligible for Septal Reduction Therapy
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Barrick SK, Greenberg MJ. Cardiac myosin contraction and mechanotransduction in health and disease. J Biol Chem. 2021;297(5):101297. https://doi.org/10.1016/j.jbc.2021.101297.
Elliott PM. The end of the beginning for drug therapy in obstructive hypertrophic cardiomyopathy with EXPLORER-HCM. Cardiovasc Res. 2020;116(13):e175–8. https://doi.org/10.1093/cvr/cvaa282.
Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424(6944):35–41. https://doi.org/10.1038/nature01780.
Park KC, Gaze DC, Collinson PO, Marber MS. Cardiac troponins: from myocardial infarction to chronic disease. Cardiovasc Res. 2017;113(14):1708–18. https://doi.org/10.1093/cvr/cvx183.
Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation. 2007;116(22):2634–53. https://doi.org/10.1161/CIRCULATIONAHA.107.187397. The first universal definition of myocardial infarction introduced a novel myocardial infarction classification system with five subcategories. These provide clinicians guidance on the interpretation of troponin levels.
Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). Eur Heart J. 2019;40(3):237–69. https://doi.org/10.1093/eurheartj/ehy462. This updated definition, following the development of more sensitive troponin assays, provides clinicians guidance on distinguishing non-ischaemic myocardial injury versus myocardial ischaemia or infarction.
Gore MO, Seliger SL, deFilippi CR, et al. Age- and sex-dependent upper reference limits for the high-sensitivity cardiac troponin T assay. J Am Coll Cardiol. 2014;63(14):1441–8. https://doi.org/10.1016/j.jacc.2013.12.032.
Giannitsis E, Mueller-Hennessen M, Zeller T, et al. Gender-specific reference values for high-sensitivity cardiac troponin T and I in well-phenotyped healthy individuals and validity of high-sensitivity assay designation. Clin Biochem. 2020;78:18–24. https://doi.org/10.1016/j.clinbiochem.2019.11.013.
Ndumele CE, Coresh J, Lazo M, et al. Obesity, subclinical myocardial injury, and incident heart failure. JACC Heart Fail. 2014;2(6):600–7. https://doi.org/10.1016/j.jchf.2014.05.017.
Klinkenberg LJJ, Wildi K, van der Linden N, et al. Diurnal rhythm of cardiac troponin: consequences for the diagnosis of acute myocardial infarction. Clin Chem. 2016;62(12):1602–11. https://doi.org/10.1373/clinchem.2016.257485.
Shave R, Baggish A, George K, et al. Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol. 2010;56(3):169–76. https://doi.org/10.1016/j.jacc.2010.03.037.
Voors AA, Tamby J-F, Cleland JG, et al. Effects of danicamtiv, a novel cardiac myosin activator, in heart failure with reduced ejection fraction: experimental data and clinical results from a phase 2a trial. Eur J Heart Fail. 2020;22(9):1649–58. https://doi.org/10.1002/ejhf.1933.
Vasile VC, Jaffe AS. The biological basis of troponin in heart disease: possible uses for troponin fragmentology. Heart Metab. 2009;43:5–8.
Chaulin AM. False-positive causes in serum cardiac troponin levels. J Clin Med Res. 2022;14(2):80–7. https://doi.org/10.14740/jocmr4664.
Saenger AK, Jaffe AS, Body R, et al. Cardiac troponin and natriuretic peptide analytical interferences from hemolysis and biotin: educational aids from the IFCC Committee on Cardiac Biomarkers (IFCC C-CB). Clin Chem Lab Med. 2019;57(5):633–40. https://doi.org/10.1515/cclm-2018-0905.
Berridge BR, Pettit S, Walker DB, et al. A translational approach to detecting drug-induced cardiac injury with cardiac troponins: consensus and recommendations from the Cardiac Troponins Biomarker Working Group of the Health and Environmental Sciences Institute. Am Heart J. 2009;158(1):21–9. https://doi.org/10.1016/j.ahj.2009.04.020.
Gheorghiade M, De Luca L, Fonarow GC, Filippatos G, Metra M, Francis GS. Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol. 2005;96(6A):11G-17G. https://doi.org/10.1016/j.amjcard.2005.07.016.
Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):e895–1032. https://doi.org/10.1161/CIR.0000000000001063.
Cardinale D, Colombo A, Sandri MT, et al. Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition. Circulation. 2006;114(23):2474–81. https://doi.org/10.1161/CIRCULATIONAHA.106.635144.
Sengupta S, Biswal S, Titus J, et al. A novel breakthrough in wrist-worn transdermal troponin-I-sensor assessment for acute myocardial infarction. Eur Heart J Digit Health. 2023;4(3):145–54. https://doi.org/10.1093/ehjdh/ztad015.
Spudich JA. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Arch. 2019;471(5):701–17. https://doi.org/10.1007/s00424-019-02259-2.
Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–21. https://doi.org/10.1126/science.aad3456.
Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106(6):1236–49. https://doi.org/10.1016/j.bpj.2014.02.011.
Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117(3):429–39. https://doi.org/10.1161/CIRCULATIONAHA.107.694158.
Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of disease progression in hypertrophic cardiomyopathy. Circ Hear Fail. 2012;5(4):535–46. https://doi.org/10.1161/CIRCHEARTFAILURE.112.967026.
Kubo T, Kitaoka H, Yamanaka S, et al. Significance of high-sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;62(14):1252–9. https://doi.org/10.1016/j.jacc.2013.03.055.
O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur Heart J. 2014;35(30):2010–20. https://doi.org/10.1093/eurheartj/eht439.
Connelly A, Coats C, Hunter A, Murday V, Findlay I. 147 Elevated serum troponin I is associated with increased risk in HCM. Heart. 2016;102(6):106–106. https://doi.org/10.1136/heartjnl-2016-309890.147.
Desai MY, Wolski K, Owens A, et al. Study design and rationale of VALOR-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy who are eligible for septal reduction therapy. Am Heart J. 2021;239:80–9. https://doi.org/10.1016/j.ahj.2021.05.007.
Cramer GE, Gommans DHF, Dieker HJ, et al. Exercise and myocardial injury in hypertrophic cardiomyopathy. Heart. 2020;106(15):1169–75. https://doi.org/10.1136/heartjnl-2019-315818.
Lampert R, Ackerman MJ, Marino BS, et al. Abstract 9544: physical activity in individuals with hypertrophic cardiomyopathy: baseline data from the prospective “Lifestyle and Exercise in HCM” (LIVE-HCM) Study. Circulation. 2021;144(1):9544–9544. https://doi.org/10.1161/circ.144.suppl_1.9544.
ACC. LIVE-HCM: vigorous exercise not associated with increased risk of cardiac events in patients with HCM. https://www.acc.org/latest-in-cardiology/articles/2023/03/01/22/45/mon-11am-live-hcm-acc-2023. Published 2023. Accessed 4 Jul 2023.
Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA. 2017;317(13):1349–57. https://doi.org/10.1001/JAMA.2017.2503.
Zamorano JL, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–79. https://doi.org/10.1093/eurheartj/ehu284. These European guidelines for hypertrophic cardiomyopathy recommend laboratory testing for troponin T, as high levels are associated with higher risk of cardiovascular events, heart failure, and death.
Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142(25):E558–631. https://doi.org/10.1161/CIR.0000000000000937. The American guidelines for hypertrophic cardiomyopathy do not provide any specific recommendations on the use of troponin levels.
McDonagh TA, Metra M, Adamo M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42(36):3599–726. https://doi.org/10.1093/eurheartj/ehab368. In the European guidelines for heart failure, initial laboratory exams recommended include troponin for exclusion of acute coronary syndrome, although elevated levels are detected in the vast majority of patients with acute heart failure.
Maron BJ, Rowin EJ, Casey SA, Maron MS. How hypertrophic cardiomyopathy became a contemporary treatable genetic disease with low mortality: shaped by 50 years of clinical research and practice. JAMA Cardiol. 2016;1(1):98–105. https://doi.org/10.1001/jamacardio.2015.0354.
Chuang C, Collibee S, Ashcraft L, et al. Discovery of aficamten (CK-274), a next-generation cardiac myosin inhibitor for the treatment of hypertrophic cardiomyopathy. J Med Chem. 2021;64(19):14142–52. https://doi.org/10.1021/acs.jmedchem.1c01290.
Malik FI, Robertson LA, Armas DR, et al. A phase 1 dose-escalation study of the cardiac myosin inhibitor aficamten in healthy participants. JACC Basic to Transl Sci. 2022;7(8):763–75. https://doi.org/10.1016/j.jacbts.2022.04.008.
Lim GB. First-in-class cardiac myosin inhibitor reduces symptoms of HCM. Nat Rev Cardiol. 2020;17(11):677. https://doi.org/10.1038/s41569-020-00453-9.
Grillo MP, Erve JCL, Dick R, et al. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first-in-class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica. 2019;49(6):718–33. https://doi.org/10.1080/00498254.2018.1495856.
Kawas RF, Anderson RL, Bartholomew Ingle SR, Song Y, Sran AS, Rodriguez HM. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem. 2017;292(40):16571–7. https://doi.org/10.1074/jbc.M117.776815.
U.S. Food and Drug Administration (FDA). FDA approves new drug to improve heart function in adults with rare heart condition. U.S. Food & Drug Administration. https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-new-drug-improve-heart-function-adults-rare-heart-condition. Published 2022. Accessed 4 Jul 2023.
Heitner SB, Jacoby D, Lester SJ, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann Intern Med. 2019;170(11):741–8. https://doi.org/10.7326/M18-3016.
Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10253):759–69. https://doi.org/10.1016/S0140-6736(20)31792-X. In EXPLORER-HCM, compared to placebo, mavacamten reduced troponin levels over 30 weeks, alongside improvements in exercise capacity, symptoms and left ventricular outflow tract obstruction.
Saberi S, Cardim N, Yamani M, et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy. Circulation. 2021;143(6):606–8. https://doi.org/10.1161/CIRCULATIONAHA.120.052359.
Desai MY, Owens A, Geske JB, et al. Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol. 2022;80(2):95–108. https://doi.org/10.1016/j.jacc.2022.04.048. In VALOR-HCM, compared to placebo, mavacamten reduced cardiac troponin-I levels over 16 weeks.
Ho CY, Mealiffe ME, Bach RG, et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;75(21):2649–2660. https://doi.org/10.1016/j.jacc.2020.03.064. In MAVERICK-HCM, compard to placebo, mavacamten resulted in rapid sustained improvements in cardiac troponin-I concentrations over 16 weeks, particularly in those with elevated levels at baseline. Amongst those with elevated baseline cardiac troponin-I concentrations, after stopping study drug at week 16, cardiac troponin-I levels increased to baseline levels by week 24.
Maron MS, Masri A, Choudhury L, et al. Phase 2 study of aficamten in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2023;81(1):34–45. https://doi.org/10.1016/j.jacc.2022.10.020. REDWOOD-HCM cohorts 1 and 2 showed that aficamten treatment was associated with non-significant reductions in troponin levels (18% relative reduction (p=0.29 compared with placebo) and 26% relative reduction (p=0.097 compared with placebo), respectively) over 10 weeks.
Cytokinetics. Cytokinetics announces results from cohort 3 of REDWOOD-HCM presented at American College of Cardiology 71st annual scientific session. Cytokinetics.https://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-announces-results-cohort-3-redwood-hcm-presented. Published 2022. Accessed June 11, 2023. REDWOOD-HCM cohort 3 showed that patients on aficamten treatment trended to lower cardiac troponin-I levels over 10 weeks.
Cytokinetics. Cytokinetics presents positive results from cohort 4 of REDWOOD-HCM and long-term results from FOREST-HCM at the American College of Cardiology 72nd annual scientific session. Cytokinetics.https://cytokineticsinc.gcs-web.com/news-releases/news-release-details/cytokinetics-presents-positive-results-cohort-4-redwood-hcm-and. Published 2023. Accessed June 11, 2023. In REDWOOD-HCM cohort 4, aficamten treatment reduced cardiac troponin-I levels at each study visit up to 10 weeks, and after the 2-week washout period, cardiac troponin-I levels returned to baseline levels. Interim analyses from FOREST-HCM also show reductions in cardiac troponin levels at 12 and 24 weeks with aficamten treatment.
Quintana E, Bajona P, Myers PO. Mavacamten for hypertrophic obstructive cardiomyopathy. Lancet. 2021;397(10272):369. https://doi.org/10.1016/S0140-6736(20)32384-9.
Psotka MA, Gottlieb SS, Francis GS, et al. Cardiac calcitropes, myotropes, and mitotropes: JACC review topic of the week. J Am Coll Cardiol. 2019;73(18):2345–53. https://doi.org/10.1016/j.jacc.2019.02.051.
He H, Baka T, Balschi J, et al. Novel small-molecule troponin activator increases cardiac contractile function without negative impact on energetics. Circ Heart Fail. 2022;15(3):e009195. https://doi.org/10.1161/CIRCHEARTFAILURE.121.009195.
Ahmad T, Miller PE, McCullough M, et al. Why has positive inotropy failed in chronic heart failure? Lessons from prior inotrope trials. Eur J Heart Fail. 2019;21(9):1064–78. https://doi.org/10.1002/ejhf.1557.
Tacon CL, McCaffrey J, Delaney A. Dobutamine for patients with severe heart failure: a systematic review and meta-analysis of randomised controlled trials. Intensive Care Med. 2012;38(3):359–67. https://doi.org/10.1007/s00134-011-2435-6.
Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325(21):1468–75. https://doi.org/10.1056/NEJM199111213252103.
Francis GS, Bartos JA, Adatya S. Inotropes. J Am Coll Cardiol. 2014;63(20):2069–78. https://doi.org/10.1016/j.jacc.2014.01.016.
Neubauer S. The failing heart-an engine out of fuel. N Engl J Med. 2007;356(11):1140–51. https://doi.org/10.1056/NEJMra063052.
Malik FI, Hartman JJ, Elias KA, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331(6023):1439–43. https://doi.org/10.1126/science.1200113.
Teerlink JR, Felker GM, McMurray JJV, et al. Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure: the ATOMIC-AHF study. J Am Coll Cardiol. 2016;67(12):1444–55. https://doi.org/10.1016/j.jacc.2016.01.031. ATOMIC-AHF reported small increases in cardiac troponin-I concentrations in patients treated with omecamtiv mecarbil compared with placebo, in the absence of other clinical evidence of myocardial ischaemia.
Teerlink JR, Felker GM, McMurray JJV, et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): a phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet. 2016;388(10062):2895–903. https://doi.org/10.1016/S0140-6736(16)32049-9. COSMIC-AHF reported small increases in cardiac troponin-I concentrations in patients treated with omecamtiv mecarbil compared with placebo, with levels returning to baseline after treatment was stopped.
Teerlink JRR, Diaz R, Felker GMM, et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Engl J Med. 2021;384(2):105–16. https://doi.org/10.1056/nejmoa2025797. GALACTIC-HF showed that cardiac troponin-I levels were higher in the omecamtiv mecarbil group compared with the placebo group. However, reassuringly, the incidence of myocardial ischaemia, ventricular arrhythmias, and death were similar in both groups with almost 7500 patient-years of follow-up.
Lewis GD, Voors AA, Cohen-Solal A, et al. Effect of omecamtiv mecarbil on exercise capacity in chronic heart failure with reduced ejection fraction: the METEORIC-HF randomized clinical trial. JAMA. 2022;328(3):259–69. https://doi.org/10.1001/jama.2022.11016.
U.S. Food & Drug Administration (FDA). December 13, 2022: Cardiovascular and Renal Drugs Advisory Committee meeting announcement. https://www.fda.gov/advisory-committees/advisory-committee-calendar/december-13-2022-cardiovascular-and-renal-drugs-advisory-committee-meeting-announcement-12132022. Published 2022. Accessed 4 Jul 2023.
U.S. Food & Drug Administration (FDA). December 13, 2022 meeting of the Cardiovascular and Renal Drugs Advisory Committee-FDA briefing document. https://www.fda.gov/media/163821/download. Published 2022. Accessed 4 Jul 2023.
Cytokinetics. Cytokinetics receives complete response letter from FDA for new drug application for omecamtiv mecarbil. Cytokinetics. https://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-receives-complete-response-letter-fda-new-drug. Published 2023. Accessed 4 Jul 2023.
Klinkenberg LJJ, van Dijk J-W, Tan FES, van Loon LJC, van Dieijen-Visser MP, Meex SJR. Circulating cardiac troponin T exhibits a diurnal rhythm. J Am Coll Cardiol. 2014;63(17):1788–95. https://doi.org/10.1016/j.jacc.2014.01.040.
Waldenström A, Ronquist G. Role of exosomes in myocardial remodeling. Circ Res. 2014;114(2):315–24. https://doi.org/10.1161/CIRCRESAHA.114.300584.
Heitner SB, Jacoby D, Lester SJ, Wang A, Zhang D, Sehnert A. Long-term safety and effectiveness of mavacamten in symptomatic obstructive hypertrophic cardiomyopathy patients, pioneer-open label extension study (PIONEER-OLE). J Am Coll Cardiol. 2019;73(9_Supplement_1):951. https://doi.org/10.1016/S0735-1097(19)31558-X.
Heitner SB, Lester S, Wang A, et al. Abstract 13962: precision pharmacological treatment for obstructive hypertrophic cardiomyopathy with mavacamten: one-year results from PIONEER-OLE. Circulation. 2019;140(1):13962–13962. https://doi.org/10.1161/circ.140.suppl_1.13962.
Saberi S, Cardim N, Yamani M, et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM cardiac magnetic resonance substudy analysis. Circulation. 2021;143(6):606–8. https://doi.org/10.1161/CIRCULATIONAHA.120.052359.
Cremer PC, Geske JB, Owens A, et al. Myosin inhibition and left ventricular diastolic function in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy: insights from the VALOR-HCM study. Circ Cardiovasc Imaging. 2022;15(12):014986. https://doi.org/10.1161/CIRCIMAGING.122.014986.
A long-term safety extension study of mavacamten (MYK-461) in adults with hypertrophic cardiomyopathy who have completed the MAVERICK-HCM (MYK-461-006) or EXPLORER-HCM (MYK-461-005) trials (MAVA-LTE). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03723655. Published 2023. Accessed 4 Jul 2023.
Florian R, Lubna C, Sara S, et al. Long-term safety of mavacamten in patients with obstructive hypertrophic cardiomyopathy: interim results of the MAVA-LONG TERM EXTENSION (LTE) study. J Am Coll Cardiol. 2021;77(18_Supplement_1):532. https://doi.org/10.1016/S0735-1097(21)01891-X.
Bristol Myers Squibb. Bristol Myers Squibb announces data from EXPLORER-LTE demonstrating sustained improvements in clinically meaningful cardiovascular outcomes at weeks 48 and 84 in patients with symptomatic obstructive hypertrophic cardiomyopathy receiving mavacamten. Bristol Myers Squibb. https://news.bms.com/news/details/2022/Bristol-Myers-Squibb-Announces-Data-from-EXPLORER-LTE-Demonstrating-Sustained-Improvements-in-Clinically-Meaningful-Cardiovascular-Outcomes-at-Weeks-48-and-84-in-Patients-with-Symptomatic-Obstructive-Hypertrophic-Cardiomyopathy-Receiving-Mavacamten/default.aspx. Published 2022. Accessed 4 Jul 2023.
Owens A, Sherrid MV, Wong TC, et al. Abstract 9685: long-term efficacy and safety of mavacamten in patients with non-obstructive hypertrophic cardiomyopathy: interim results from the MAVERICK-LTE cohort of the MAVA-LTE study. Circulation. 2021;144(1):9685–9685. https://doi.org/10.1161/circ.144.suppl_1.9685.
Tian Z, Wang F, Jin W, et al. Study design and rationale of EXPLORER-CN: a phase III, randomised, double-blind, placebo-controlled clinical study to evaluate the efficacy and safety of mavacamten in Chinese adults with symptomatic obstructive hypertrophic cardiomyopathy. BMJ Open. 2023;13(6):e071473. https://doi.org/10.1136/bmjopen-2022-071473.
Squibb B-M. A study of mavacamten in non-obstructive hypertrophic cardiomyopathy (ODYSSEY-HCM). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05582395. Published 2022. Accessed 4 Jul 2023.
CY 6022 is an open label study to collect long-term safety and tolerability data for aficamten (CK-3773274) (FOREST-HCM). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04848506. Published 2023. Accessed 4 Jul 2023.
Cytokinetics. Cytokinetics announces data from REDWOOD-HCM OLE and GALACTIC-HF presented as late breaking science presentations at the European Society of Cardiology heart failure 2022 congress. Cytokinetics. https://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-announces-data-redwood-hcm-ole-and-galactic-hf. Published 2022. Accessed 4 Jul 2023.
Cytokinetics. Cytokinetics presents new data from REDWOOD-HCM OLE in late breaking clinical trial session at the HFSA annual scientific meeting. Cytokinetics. https://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-presents-new-data-redwood-hcm-ole-late-breaking-0. Accessed 4 Jul 2023.
CY 6031 study will evaluate the effects of treatment with aficamten (CK-3773274) over a 24-week period on cardiopulmonary exercise capacity and health status in patients with symptomatic oHCM (SEQUOIA-HCM). ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT05186818. Published 2022. Accessed 4 Jul 2023.
Cytokinetics. The purpose of this study is to compare the efficacy and safety of aficamten (CK-3773274) compared with metoprolol succinate in adults with symptomatic hypertrophic cardiomyopathy and left ventricular outflow tract obstruction (MAPLE-HCM). ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT05767346. Published 2023. Accessed 4 Jul 2023.
Teerlink JR, Clarke CP, Saikali KG, et al. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first-in-man study. Lancet. 2011;378(9792):667–75. https://doi.org/10.1016/S0140-6736(11)61219-1.
Greenberg BH, Chou W, Saikali KG, et al. Safety and tolerability of omecamtiv mecarbil during exercise in patients with ischemic cardiomyopathy and angina. JACC Heart Fail. 2015;3(1):22–9. https://doi.org/10.1016/j.jchf.2014.07.009. Erratum in: JACC Heart Fail. 2020;8(8):700.
Cleland JGF, Teerlink JR, Senior R, et al. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet. 2011;378(9792):676–83. https://doi.org/10.1016/S0140-6736(11)61126-4.
Cytokinetics. Safety, PK, and efficacy of omecamtiv mecarbil in Japanese subjects with heart failure with reduced ejection fraction. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/history/NCT02695420. Published 2021. Accessed 4 Jul 2023.
Author information
Authors and Affiliations
Contributions
M.M.Y.L. and A.M. wrote the main manuscript text. M.M.Y.L. prepared figures 1-2. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Lee received research grants through his institution, the University of Glasgow, from AstraZeneca, Boehringer Ingelheim and Roche Diagnostics; is a member of a Trial Steering Committee for Cytokinetics and Clinical Endpoints Committee for Bayer.
Masri received research grants from Ionis, Cytokinetics, and Pfizer; and fees (honoraria or consulting) from Cytokinetics, BMS, Eidos, Pfizer, Ionis, Alnylam, Attralus, Haya, BioMarin, Lexicon, and Tenaya.
Human and Animal Rights
All reported studies with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards and international/national/institutional guidelines).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: correction in Figure 2, removal of [87], [88] and [88] in Tables 1, 3 and 4 captions and order of references 78-88
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, M.M.Y., Masri, A. Differentiating Cardiac Troponin Levels During Cardiac Myosin Inhibition or Cardiac Myosin Activation Treatments: Drug Effect or the Canary in the Coal Mine?. Curr Heart Fail Rep 20, 504–518 (2023). https://doi.org/10.1007/s11897-023-00620-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-023-00620-2