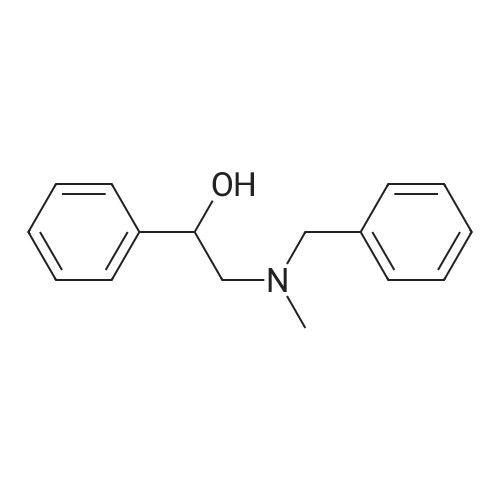
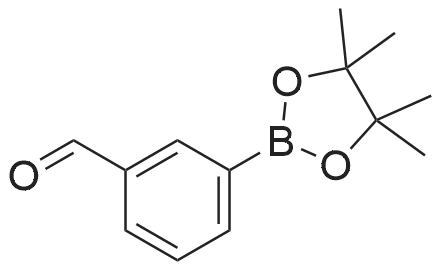
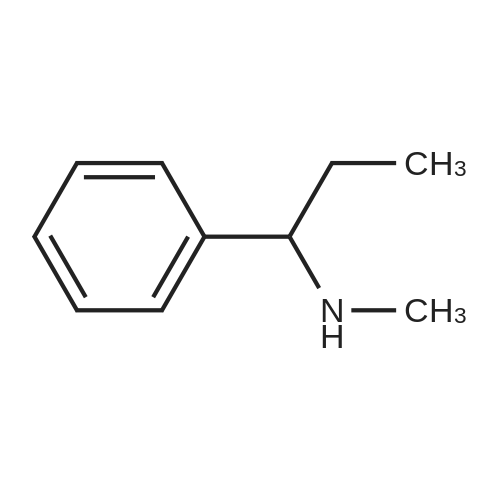
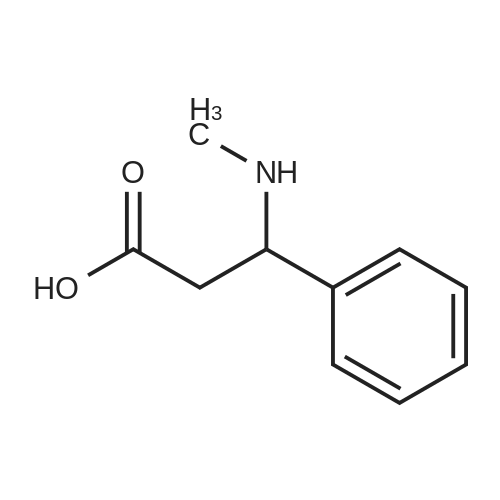
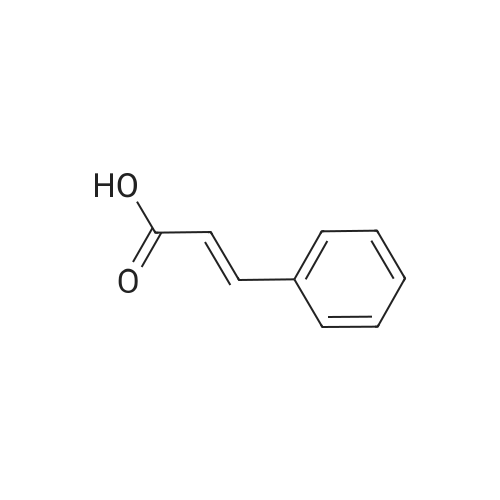
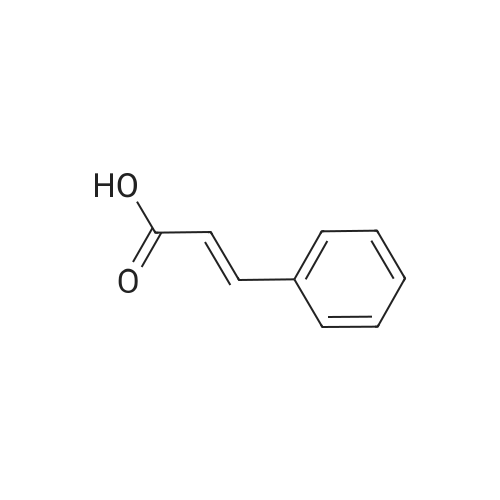
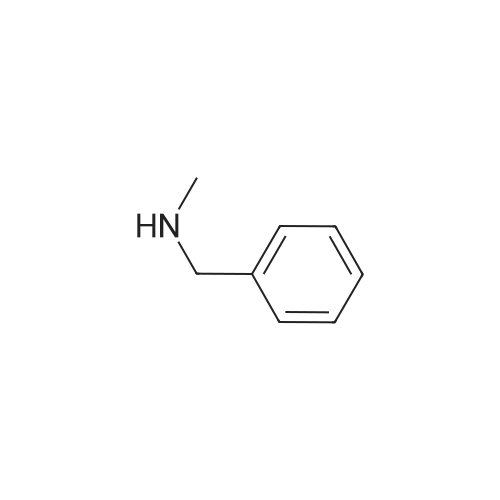
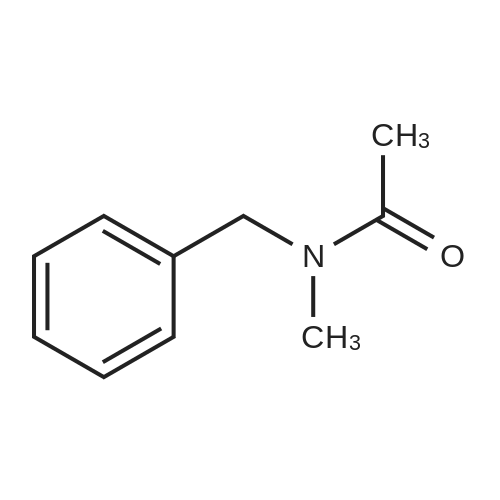
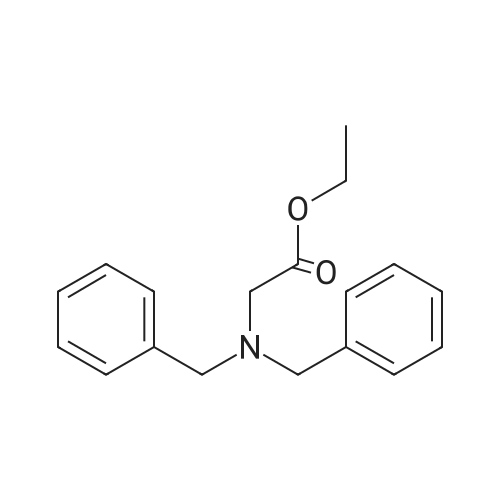
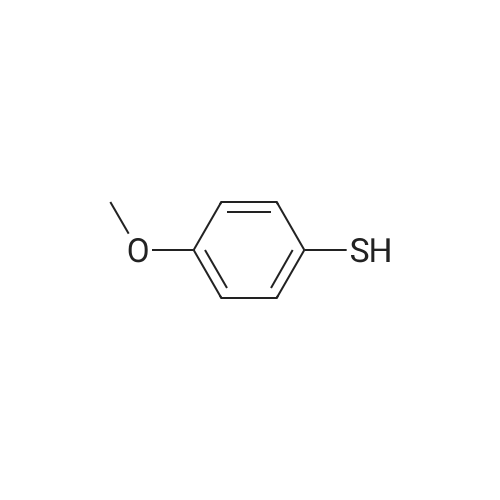
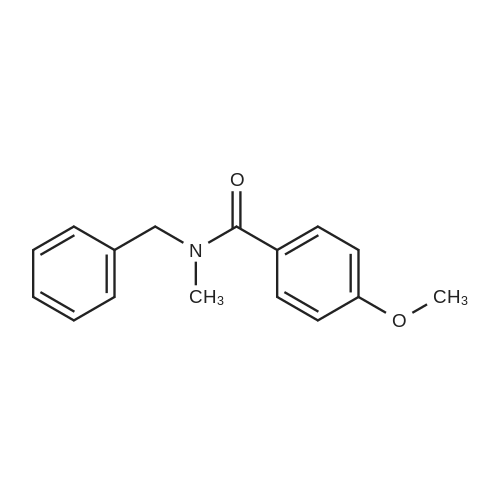
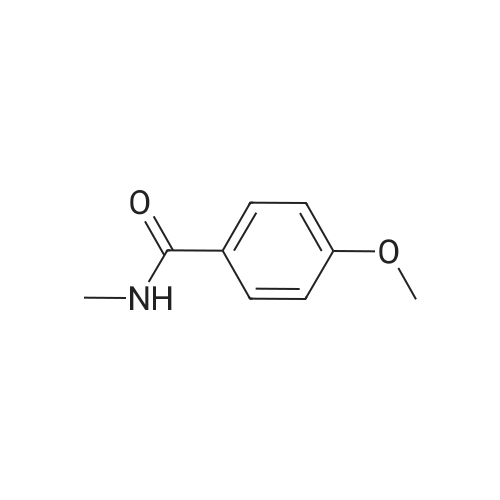
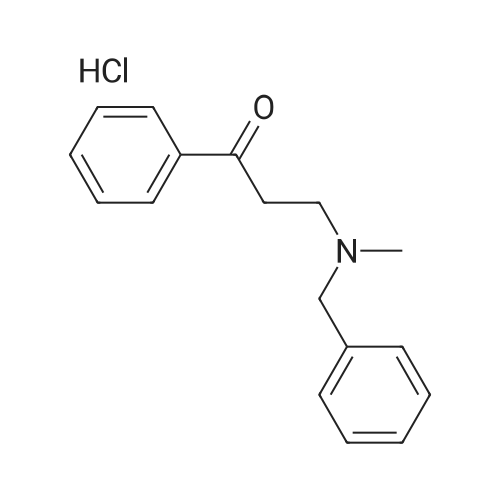
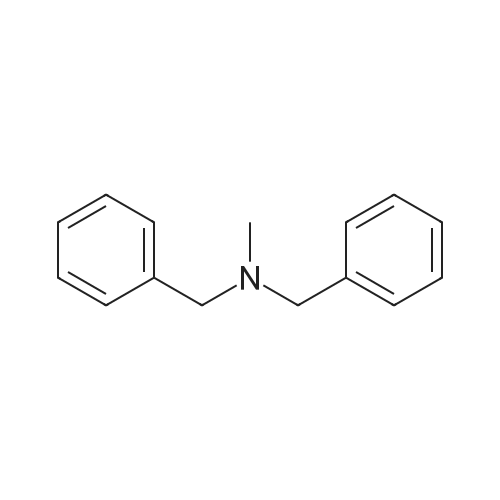
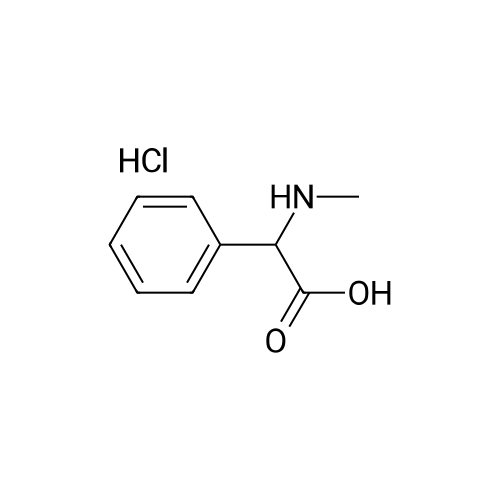
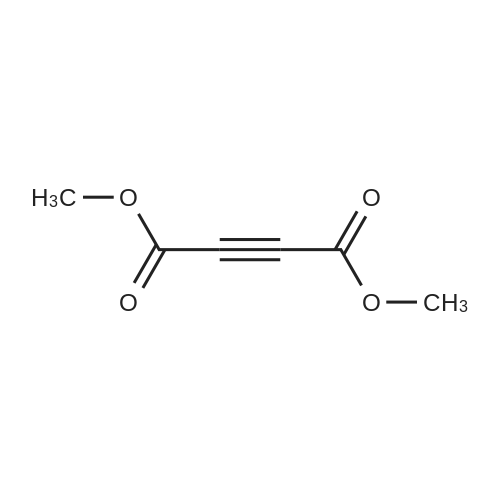
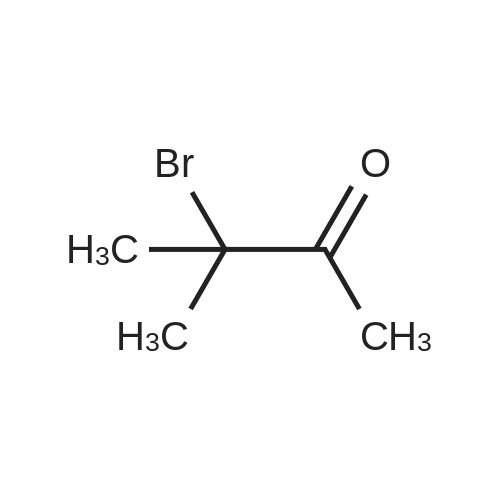
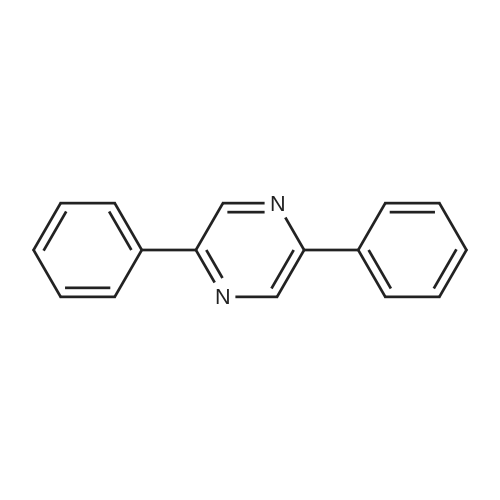
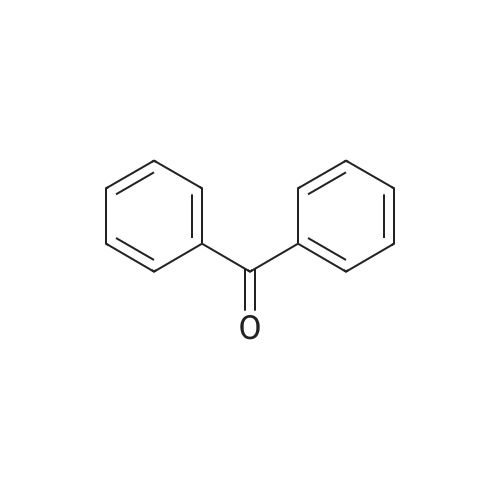
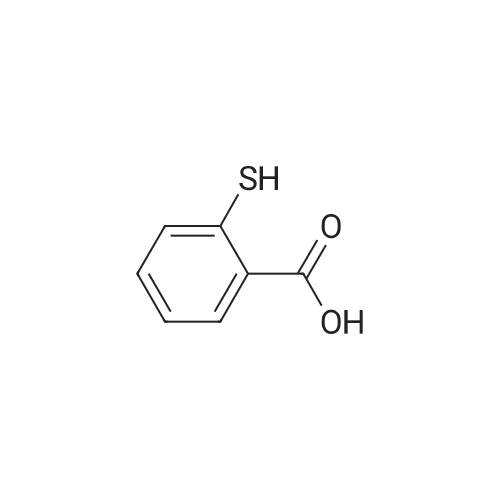
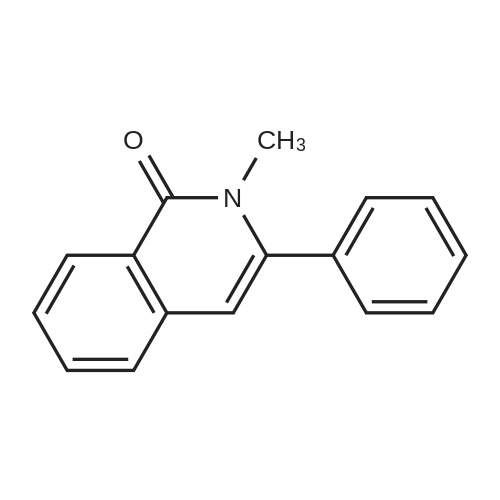
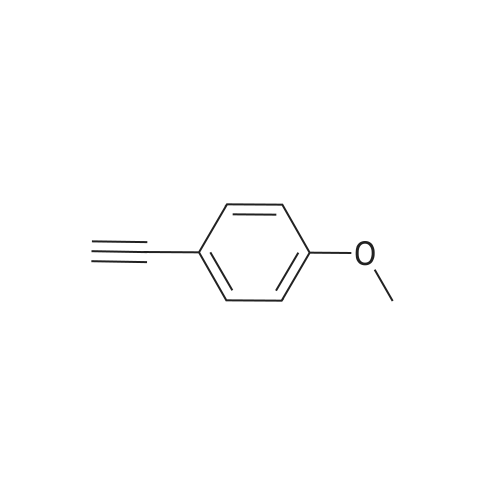
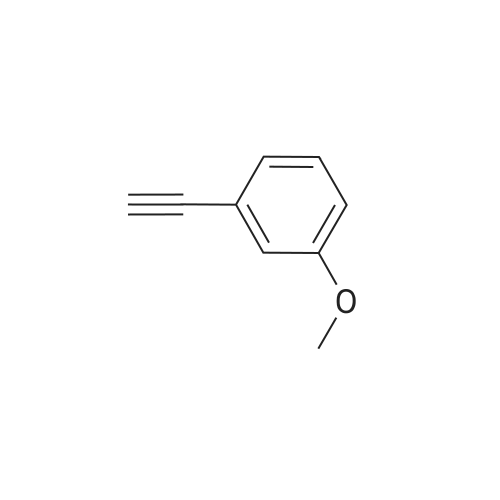
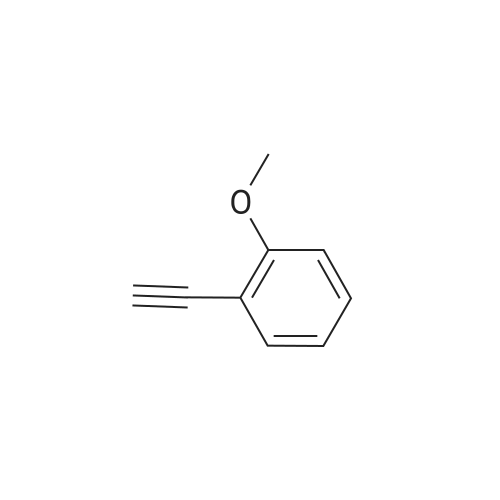
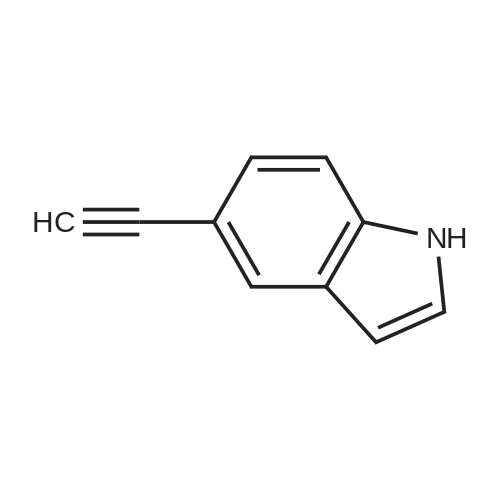
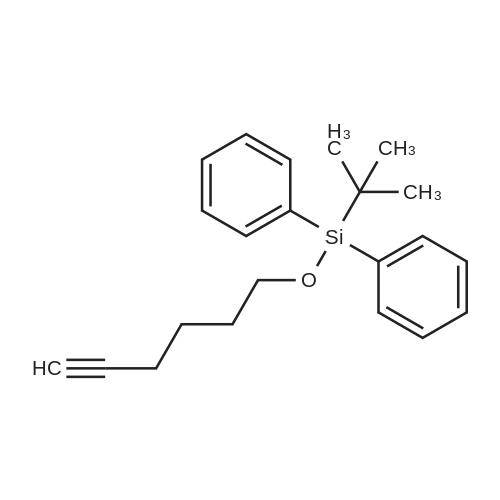
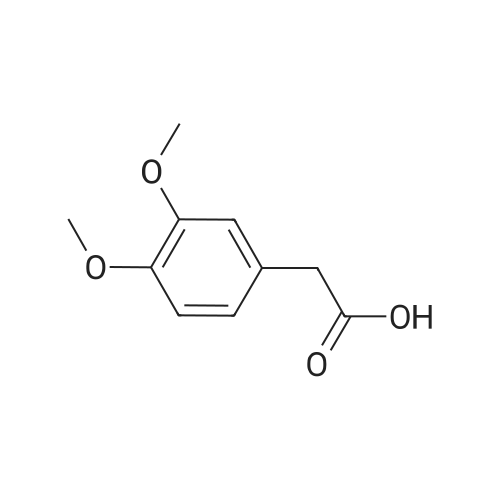
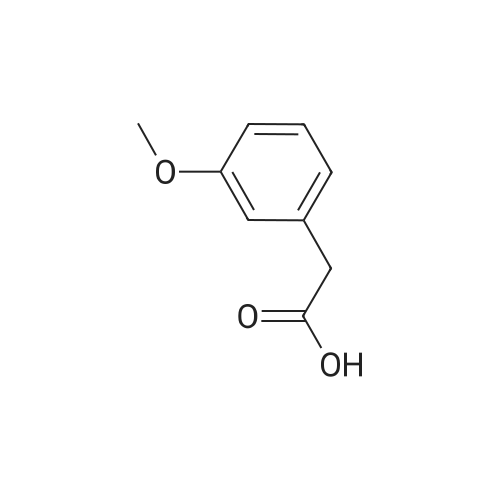
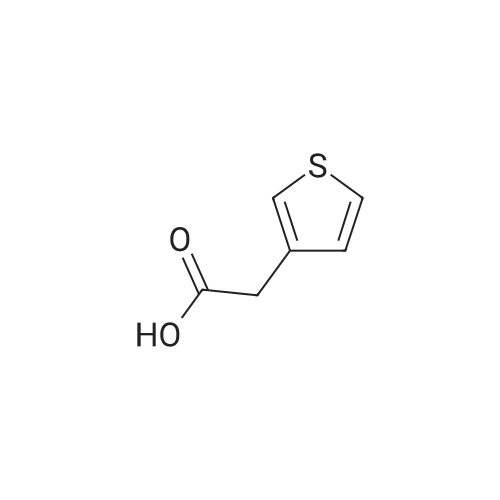
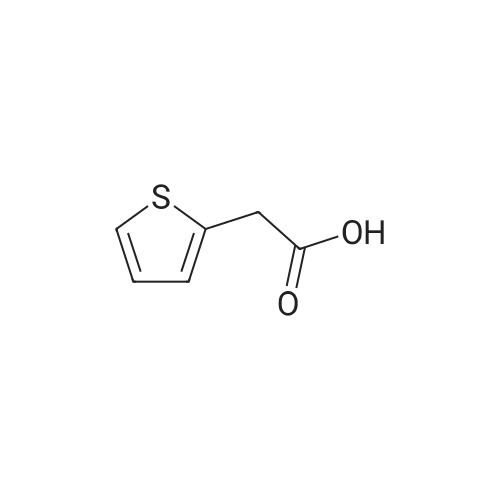
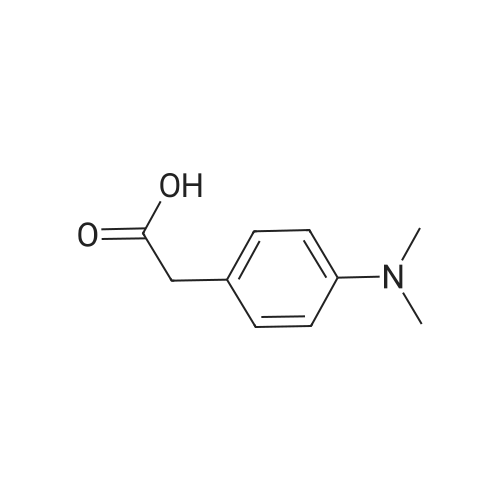
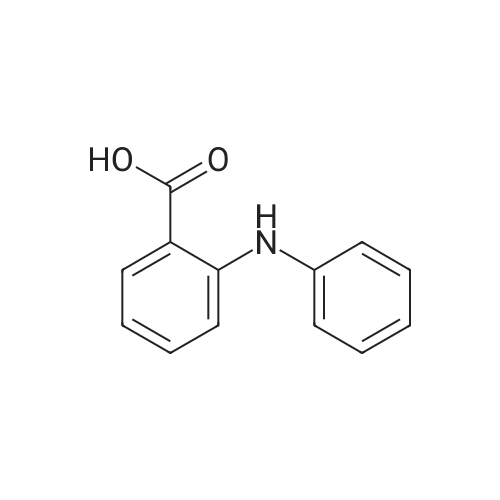
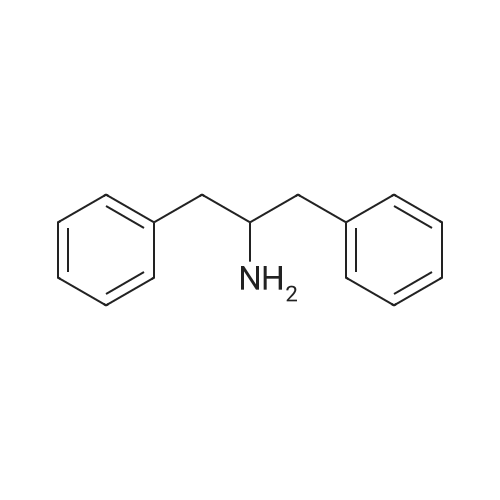
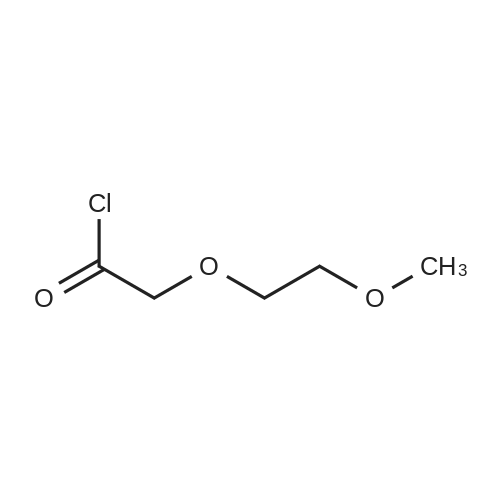
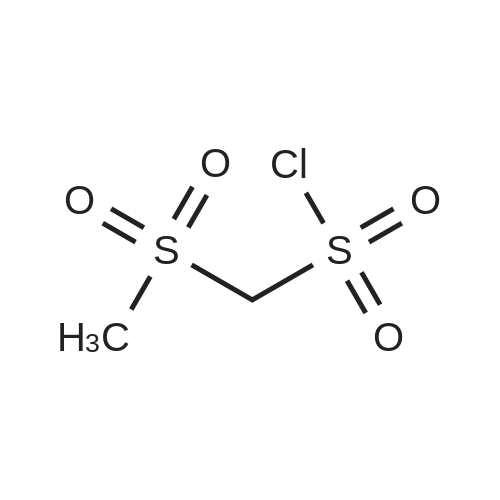
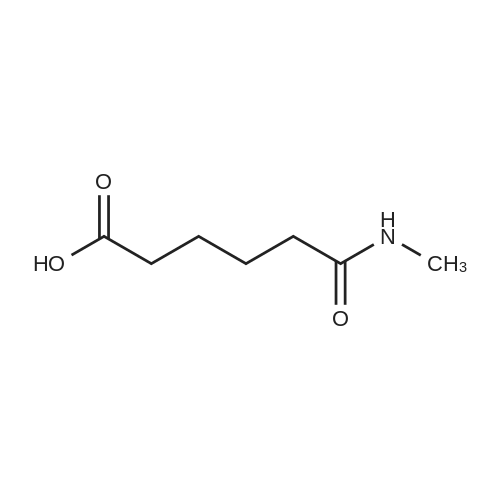
| 96% |
With tetrakis[3,5-bis(trifluoromethyl)phenyl]boric acid bis(diethyl ether) complex; (PNP<SUP>Cy</SUP>)Co(CH<SUB>2</SUB>SiMe<SUB>3</SUB>); isopropanol In tetrahydrofuran at 80℃; for 24h; Inert atmosphere; Glovebox; Schlenk technique; Sealed tube; |
|
| 95% |
With hydrogen; tri-isobutyl aluminium In toluene at 100℃; for 24h; |
|
| 92% |
With hydrogen In toluene at 90℃; for 96h; other catalysts; reaction in the presence of PhSiH3; organolanthanide-catalyzed imine hydrogenation; selectivity; mechanistic observations; stoichiometric reaction with Sm complex; |
|
| 89% |
With 1,2-bis[(2,6-diisopropylphenyl)imino]acenaphthenecobalt(II) dibromide; hydrogen; lithium triethylhydroborate In tetrahydrofuran at 60℃; for 24h; |
|
| 88% |
With tetrakis[3,5-bis(trifluoromethyl)phenyl]boric acid bis(diethyl ether) complex; C32H63CoNP2Si; hydrogen In tetrahydrofuran at 60℃; for 72h; |
|
| 88% |
With tetrakis[3,5-bis(trifluoromethyl)phenyl]boric acid bis(diethyl ether) complex; (PNP<SUP>Cy</SUP>)Co(CH<SUB>2</SUB>SiMe<SUB>3</SUB>); hydrogen In tetrahydrofuran at 60℃; for 72h; |
In a typical experiment, complex 1 (6.1 mg, 10 tmol) and H[BAr’4].(Et2O)2 (10.1 mg, 10 tmol) were dissolved in THF (2.0 mE) in a 100 mE thick-walled glass vessel equipped with a TEFLON stopcock and a stir bar. The substrate (0.5 mmol) to be hydrogenated was then added. The vessel was degassed by freeze-pump-thaw and then hydrogen (1 or 4 atm) was added. The resulting solution was stirred at the desired temperature (25-60° C.) for the indicated reaction time. At the end of the reaction, the solvent was evaporated and the residue was passed through silica gel in a pipette. The solvent was removed under vacuum and the ‘H NMR spectrum of the crude product mixture was recorded in CDC13. Hydrogenation products were then isolated by column chromatography or preparative thin layer chromatography (“TLC”) using n-hexane/ethyl acetate (3:1, v/v) as an eluent. Isolated products were characterized by ‘H NMR and GCMS, with spectra matching those reported in the literature or authentic samples. |
| 86% |
With sodium tetrahydridoborate In methanol at 5℃; |
|
| 83% |
With C23H25IIrN3; hydrogen In 2,2,2-trifluoroethanol at 35℃; for 6h; |
|
| 80.1% |
With BH3 In methanol for 0.5h; Ambient temperature; |
|
| 80% |
With lithium aluminium hydride In tetrahydrofuran; diethyl ether for 16h; Heating; |
|
| 79% |
With hydrogen; C44H62AlLiN2O2 In toluene at 100℃; for 24h; Sealed tube; |
|
| 65% |
With dimethylsulfide borane complex In chloroform-d1 at 60℃; for 30h; Sealed tube; Schlenk technique; chemoselective reaction; |
General procedure for reduction of imines
General procedure: After 1.0 mmol of an imine (entries 1-12) and Me2S-BH3 (amounts according to Table 1) were mixed in a sealed J. Young NMR tube using about 1 mL of CDCl3, the reaction mixture was left at 60 °C for the time duration indicated in Table 1. For α,β-unsaturated imines (entries 13 and 14), the reactants were mixed in CH2Cl2 at -78 °C. After the reaction was completed (via 1H-NMR spectroscopy), it was quenched with 5 mL of MeOH, followed by removal of all volatiles under reduced pressure. The crude product mixture was then dissolved in 10 mL ethyl acetate, washed three times with 10 mL of water/brine, and dried with MgSO4. All amine samples were collected as oils after removal of solvent apart from benzylmethylamine (entry 1) and N-benzylaniline (entry 4), which were obtained as solids. The spectroscopic data for all amines matched those reported (Table 2). |
| 38% |
With hydrogen; gold In toluene at 50℃; for 24h; Autoclave; |
|
|
With nickel at 100℃; Hydrogenation; |
|
|
With nickel at 75℃; Hydrogenation; |
|
|
With ethanol; nickel at 25℃; Hydrogenation; |
|
|
With sodium mercury amalgam; ethanol |
|
|
With ethanol durch elektrolytische Reduktion; |
|
|
With anhydrous Sodium acetate durch elektrolytische Reduktion an einer Blei-Kathode; |
|
|
With lithium aluminium hydride In diethyl ether for 2h; Heating; |
|
|
With tris(triphenylphosphine)ruthenium(II) chloride; potassium carbonate; isopropanol for 9h; Heating; |
|
|
With phenylsilane; hydrogen In toluene at 90℃; for 44h; |
|
|
With diphenylsilane for 24h; Ambient temperature; |
|
|
With indium powder; ammonia hydrochloride In ethanol for 2.5h; Heating; |
|
|
With sodium tetrahydridoborate |
|
|
With sodium tetrahydridoborate In methanol; lithium hydroxide monohydrate at 20℃; for 1h; |
|
|
With hydrogen |
|
|
With sodium tetrahydridoborate; toluene-4-sulfonic acid at 25℃; for 0.166667h; |
|
|
With sodium tetrahydridoborate |
|
|
Multi-step reaction with 2 steps
1: sulfur / 80 - 90 °C / im Rohr
2: amalgamated aluminium; aqueous alcohol |
|
| 4 %Spectr. |
With hydrido[bis(pentafluorophenyl)]{2-[(2,2,6,6-tetramethylpiperidinium-1-yl)methyl]phenyl}borate; hydrogen In toluene at 110℃; for 24h; Inert atmosphere; |
|
|
With sodium tetrahydridoborate In ethanol at 20℃; for 4h; |
|
|
With sodium tetrahydridoborate In ethanol |
|
|
With [Ir(NCCH3)(1,5-cyclooctadiene)((1-methylimidazolyl)(2-methoxybenzyl))][BF4]; isopropanol; potassium hydroxide at 80℃; for 5.75h; Inert atmosphere; chemoselective reaction; |
|
|
With sodium tetrahydridoborate In methanol; lithium hydroxide monohydrate at 0 - 20℃; for 2.5h; |
General procedure: m-Tolualdehyde (1.66 g, 14.0 mmol) was treated with methylamine (40% aqueous, 1.39 ml, 18.0 mmol) in methanol (20 ml) at room temperature. The reaction was stirred for 20 min and treated with sodium borohydride (0.26 g, 7.0 mmol) portionwise. The reaction was stirred for 1 h and treated with 3′-fluoro-2-bromoacetophenone (3.0 g, 14.0 mmol) followed by stirring for 45 min at room temperature. The reaction was finally treated with sodium borohydride (0.52 g, 14.0 mmol) portionwise, and stirring continued overnight. The reaction was diluted with water (100 ml) and extracted with methylene chloride (3 × 100 ml). The combined organic extracts were washed with brine and dried over anhydrous sodium sulfate, followed by filtration and concentration in vacuo. Purification by column chromatography on silica gel eluting with hexanes/ethyl acetate (3/1) provided the amino alcohol (4.3 g) as a yellow oil |
|
With [(η6-C6H6)Ru(6,6’-(OH)2-bpy)Cl]Cl; hydrogen; triethylamine In tetrahydrofuran for 48h; Reflux; |
2 Example 2
One exemplary catalyst according to present invention represented by formula: was used for hydrogenation of several different organic substrates. For ketones, the reaction was carried out by mixing 1 mole% of catalyst, 10:90 of MeOH: water and Na02CH, and heated to reflux for 16-18 hours. For imines, the reaction was carried out by mixing 1 mole% of catalyst, THF, NEt3 base (5 molar %) and 75 psi (g), with heating to reflux for 48 hours. The catalyst provided a very high percentage conversion for ketones in water. The results are shown in Table 2. |
| 99 %Chromat. |
With propane; hydrogen at 40 - 50℃; Supercritical conditions; Flow reactor; |
|
|
Multi-step reaction with 2 steps
1: dicarbonyl(cyclopentadienyl)methyliron(II) / hexadeuterobenzene / 24 h / 80 °C / Sealed tube; Inert atmosphere
2: dicarbonyl(cyclopentadienyl)methyliron(II); triethylsilane |
|
| > 99 %Spectr. |
With poly[3-oxydiethylenediamine-(1,4-benzene)bisborane] In tetrahydrofuran-d8 at 40℃; for 12h; Inert atmosphere; Sealed tube; |
|
|
With C23H24IN3Ru; hydrogen In 2,2,2-trifluoroethanol at 40℃; for 12h; |
|
|
With ethanol durch elektrolytische Reduktion; |
|
| 84 %Chromat. |
With borane-ammonia complex; [K(thf)1.5{(2,6-diisopropylphenyl bisaryl-(imino)acenaphthene)Co(η4-1,5-cyclooctadiene)}] In tetrahydrofuran at 25℃; for 6h; Inert atmosphere; Sealed tube; |
|
| 88 %Chromat. |
Stage #1: N-Benzylidenemethylamine With lithium triethylhydroborate; cobalt(II) dibromide In tetrahydrofuran Inert atmosphere; Glovebox;
Stage #2: With hydrogen In tetrahydrofuran at 60℃; for 24h; |
|
| 65 %Chromat. |
With 1,4-diaza-bicyclo[2.2.2]octane; anhydrous silver tetrafluoroborate; C15H4BClCoF18N6; hydrogen In tetrahydrofuran at 60℃; for 10h; |
|
| > 95 %Spectr. |
With hydrogen; C24H45NP2Zn In toluene at 120℃; for 18h; Autoclave; Green chemistry; |
|
|
With sodium tetrahydridoborate In methanol at 20℃; for 16h; Cooling with ice; |
|
|
With sodium tetrahydridoborate In methanol at 20℃; for 1h; Cooling with ice; |
Step 1
General procedure: To a solution of aldehyde (1.0 eq) and alkylamine (1.1 eq) in dry DCM was added Na2SO4 and stirred overnight. The mixture was filtered and evaporated under reduced pressure. Then, the residue was re-dissolved in methanol, carefully added NaBH4 (0.5 eq) under ice bath and was stirred for 1 h at room temperature. The reaction was quenched by saturated Na2CO3, extracted with EA and dried over Na2SO4. The combined organic phase was evaporated to afford the desired intermediate 16. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping