Peroxisomes in Immune Response and Inflammation


 , ,
, ,  ,
,
Abstract
:1. Introduction
2. The Regulation of Peroxisomal Genes is Correlated to Proliferation and Activation of Various Immune Cells
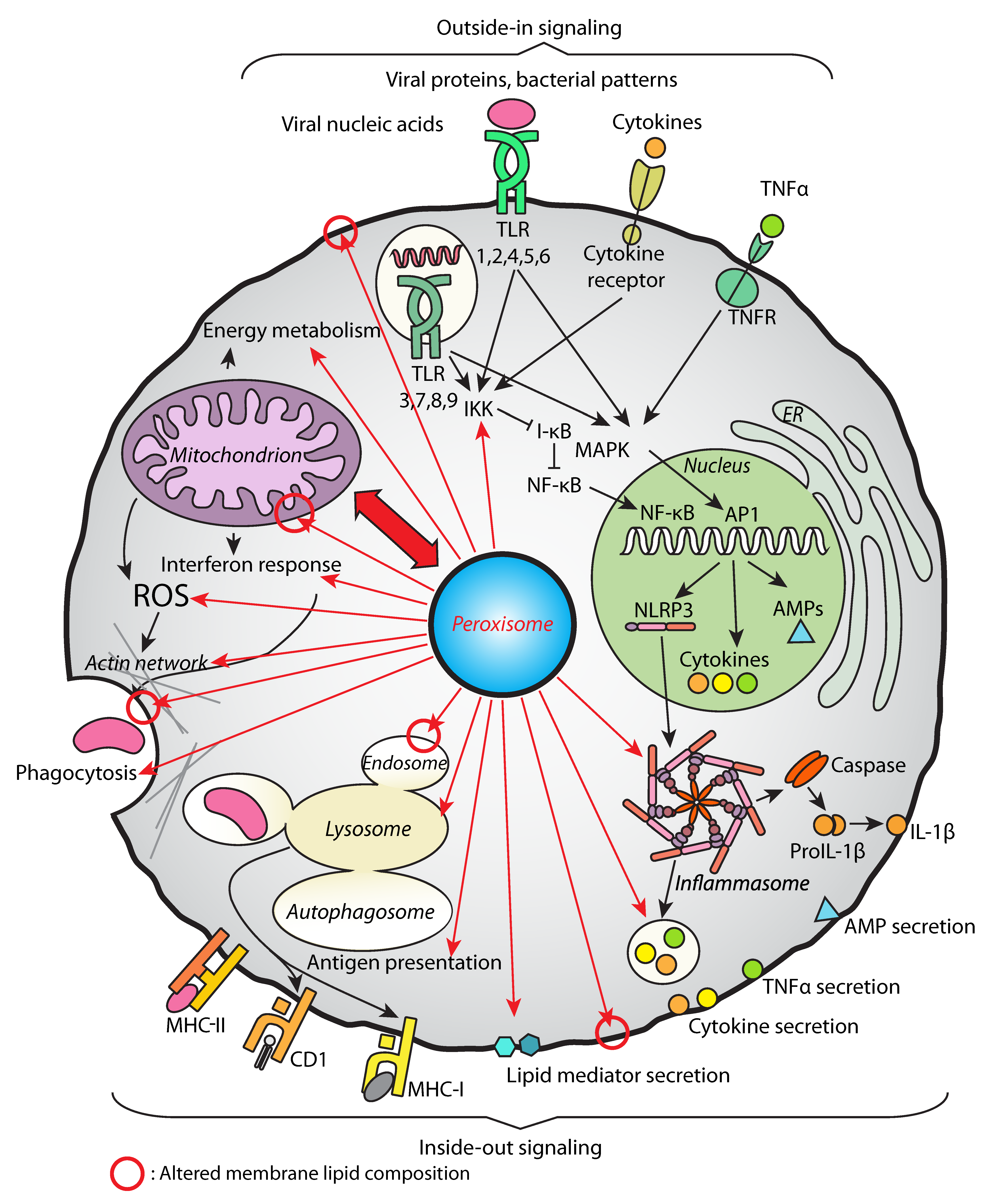
3. Peroxisomes Orchestrate the Immune Response Strategies during Systemic Infection
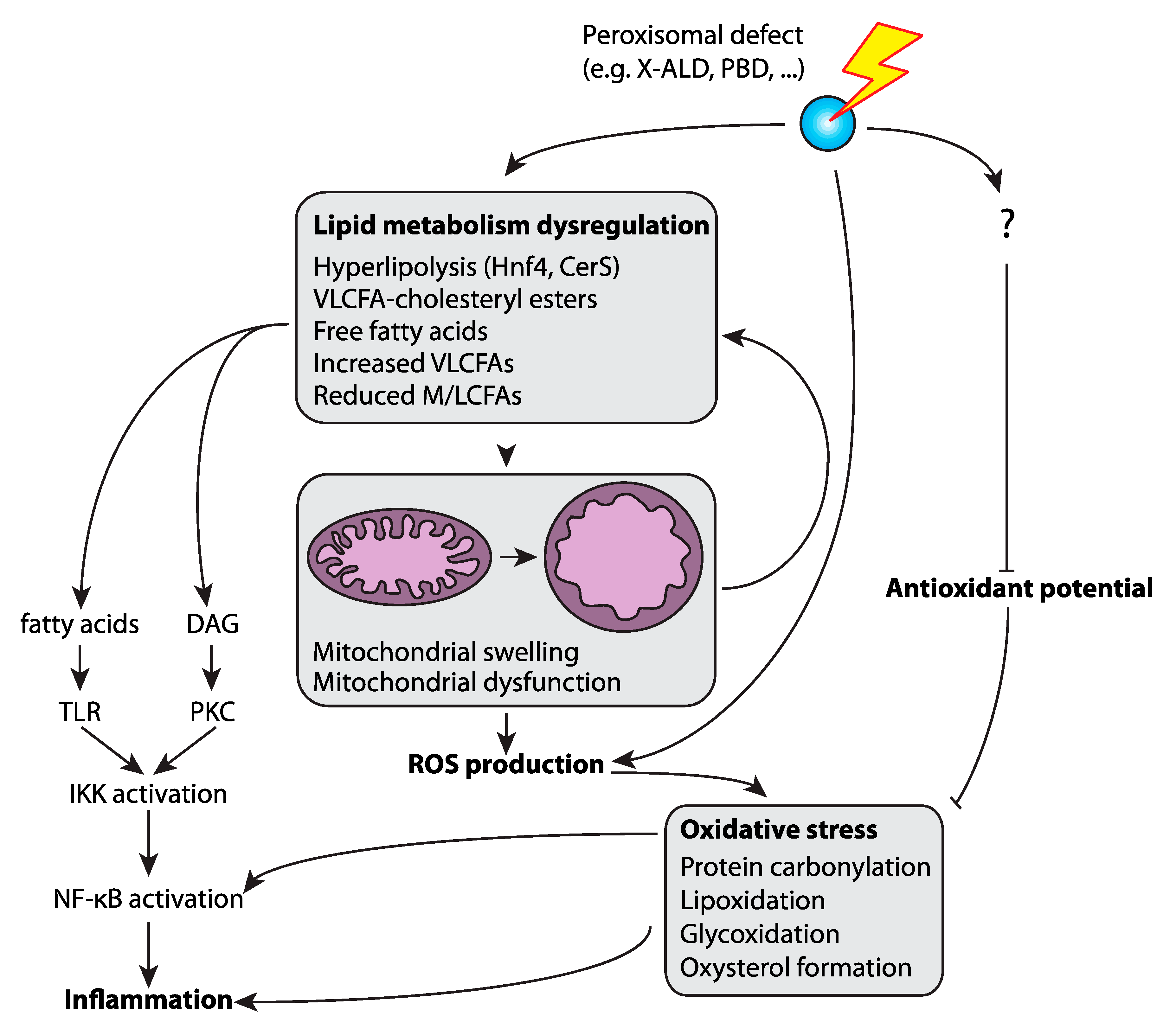
4. Peroxisome Metabolism Protects from Neuroinflammation
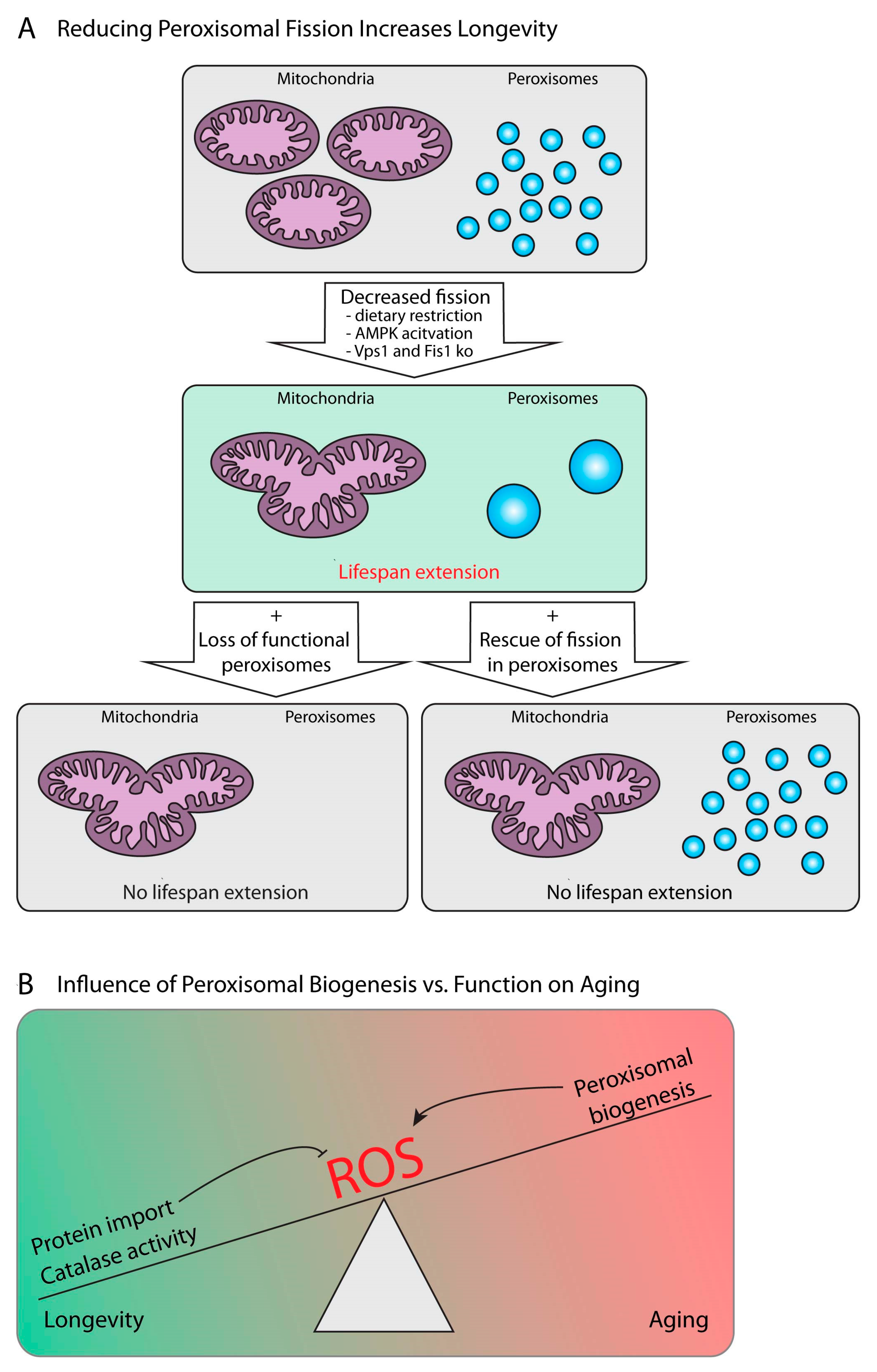
5. Peroxisome Metabolism during Aging
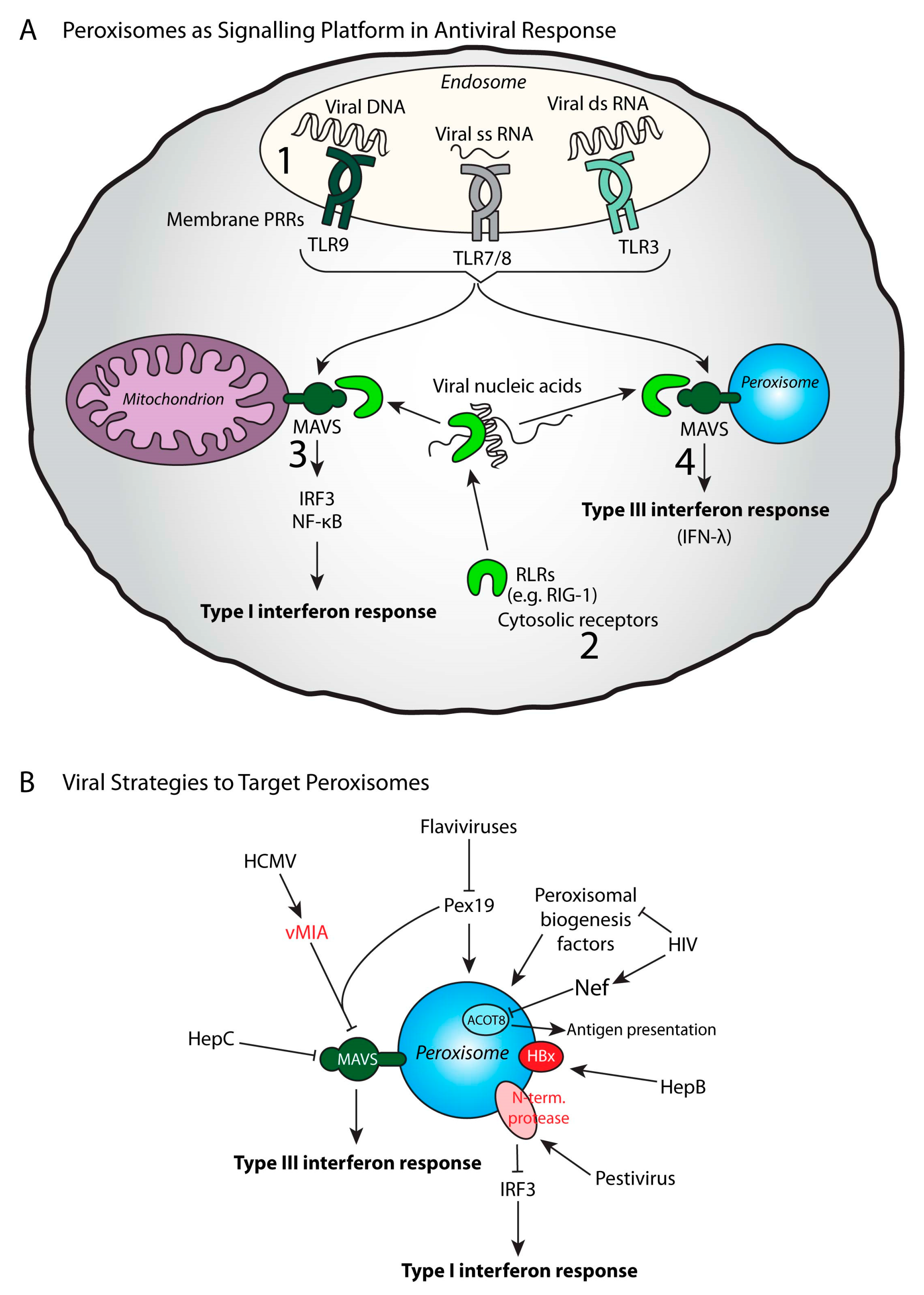
6. Peroxisomes Act as Signaling Platforms to Activate Antiviral Responses
7. Peroxisomal Functions are Linked to Phagocytosis Capacity
8. Conclusion and Future Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AASA | Aminoadipic semialdehyde |
| ABC | ATP-binding Cassette |
| ACOT-8 | Acyl-coenzyme A thioesterase 8 |
| ACOX1 | Acyl-coenzyme A oxidase 1 |
| AD | Alzheimer’s disease |
| AMN | Adrenomyeloneuropathy |
| AMPK | Adenosine monophosphate-activated protein kinase |
| AMPs | Antimicrobial peptides |
| cALD | Cerebral adrenoleukodystrophy |
| CD | Cluster of differentiation |
| CEL | Carboxyethyl-lyine |
| CerS | Ceramide synthase |
| COX | Cyclooxygenase |
| CNS | Central nervous system |
| DHA | Docosahexaenoic acid, C22:6 n-3 |
| DAG | Diacyl glycerol |
| DPA | Docosapentaenoic acid, C22:5 n-3 |
| dsRNA | Double-stranded RNA |
| EPA | Eicosapentaenoic acid, C20:5 n-3 |
| Fis1 | Fission 1 protein |
| GPX | Glutathion peroxidase |
| GSA | Glutamic semialdehyde |
| HCMV | Human cytomegalovirus |
| HepB/C | Hepatitis B or C virus |
| HIV | Human immunodeficiency virus |
| Hnf4 | Hepatonuclear factor 4 |
| IFN | Interferon |
| I-κB | Inhibitor of NF-κB |
| IKK | I-κB kinase |
| IL | Interleukin |
| iNKT | invariant natural killer T cells |
| IPF | Idiopathic pulmonary fibrosis |
| LCFAs | Long-chain fatty acids |
| LPS | Lipopolysaccharide |
| MDAL | Malonedialdehyde-lysine |
| MAPK | Mitogen-activated protein kinase |
| MAVS | Mitochondrial antiviral signaling adaptor |
| MCFAs | Medium-chain fatty acids |
| MFP-2 | Multifunctional protein-2 |
| MHC | Major histocompatibility complex |
| MS | Multiple sclerosis |
| MUFAs | Monounsaturated fatty acids |
| Nef | Negative regulatory factor |
| NF-κB | Nuclear Factor kappa-light-chain- -enhancer of activated B cells |
| NK | Natural killer |
| NO | Nitric oxide |
| PBD | Peroxisome biogenesis disorder |
| PBMC | Peripheral blood mononuclear cells |
| PEX | Peroxin |
| PKC | Protein kinase C |
| PRRs | Pattern recognition receptors |
| PTS | Peroxisome targeting signal |
| PUFAs | Polyunsaturated fatty acids |
| RIG-1 | Retinoic acid inducible gene 1 |
| RLRs | RIG-I-like receptors |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| ssRNA | Single stranded RNA |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| VLCFAs | Very long-chain fatty acids |
| vMIA | Viral mitochondria-localized inhibitor of apoptosis |
| Vps1 | Vacuolar protein sorting-associated protein |
| X-ALD | X-linked Adrenoleukodystrophy |
References
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Peroxisomes and Their Central Role in Metabolic Interaction Networks in Humans. Subcell. Biochem. 2018, 89, 345–365. [Google Scholar] [PubMed]
- Shai, N.; Yifrach, E.; van Roermund, C.W.T.; Cohen, N.; Bibi, C.; Jlst, L.I.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, W.; Shi, B.; Klein, S.; Su, X. Peroxisomal regulation of redox homeostasis and adipocyte metabolism. Redox Biol. 2019, 24, 101167. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Bonekamp, N.A.; Islinger, M. Fission and proliferation of peroxisomes. Biochim. Biophys. Acta 2012, 1822, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, A.; Tiwari-Pandey, R.; Pandey, N.R. Mitochondria: The indispensable players in innate immunity and guardians of the inflammatory response. J. Cell Commun. Signal. 2019. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Vamecq, J.; Cherkaoui-Malki, M.; Andreoletti, P.; Latruffe, N. The human peroxisome in health and disease: The story of an oddity becoming a vital organelle. Biochimie 2014, 98, 4–15. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, N.A.; Volkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. Biofactors 2009, 35, 346–355. [Google Scholar] [CrossRef]
- Diczfalusy, U.; Kase, B.F.; Alexson, S.E.; Bjorkhem, I. Metabolism of prostaglandin F2 alpha in Zellweger syndrome. Peroxisomal beta-oxidation is a major importance for in vivo degradation of prostaglandins in humans. J. Clin. Investig. 1991, 88, 978–984. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Mayatepek, E.; Keppler, D. Peroxisomal leukotriene degradation: Biochemical and clinical implications. Adv. Enzym. Regul. 1993, 33, 181–194. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Meissner, T.; Wanders, R.J.; Mayatepek, E. Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of leukotrienes. Biochem. Biophys. Res. Commun. 2002, 293, 269–273. [Google Scholar] [CrossRef]
- Giordano, C.R.; Terlecky, S.R. Peroxisomes, cell senescence, and rates of aging. Biochim. Biophys. Acta 2012, 1822, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Mooijer, P.A.; Zhang, Z.; Reddy, J.K.; Spector, A.A.; Wanders, R.J. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J. Lipid Res. 2001, 42, 1987–1995. [Google Scholar]
- Sprecher, H. The roles of anabolic and catabolic reactions in the synthesis and recycling of polyunsaturated fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2002, 67, 79–83. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef]
- Dowds, C.M.; Kornell, S.C.; Blumberg, R.S.; Zeissig, S. Lipid antigens in immunity. Biol. Chem. 2014, 395, 61–81. [Google Scholar] [CrossRef]
- Hubler, M.J.; Kennedy, A.J. Role of lipids in the metabolism and activation of immune cells. J. Nutr. Biochem. 2016, 34, 1–7. [Google Scholar] [CrossRef]
- Puertollano, M.A.; de Pablo, M.A.; Alvarez de Cienfuegos, G. Immunomodulatory effects of dietary lipids alter host natural resistance of mice to Listeria monocytogenes infection. FEMS Immunol. Med. Microbiol. 2001, 32, 47–52. [Google Scholar] [CrossRef]
- Sadik, C.D.; Luster, A.D. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012, 91, 207–215. [Google Scholar] [CrossRef]
- Yaqoob, P. Lipids and the immune response: From molecular mechanisms to clinical applications. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 133–150. [Google Scholar] [CrossRef]
- Vijayan, V.; Srinu, T.; Karnati, S.; Garikapati, V.; Linke, M.; Kamalyan, L.; Mali, S.R.; Sudan, K.; Kollas, A.; Schmid, T.; et al. A New Immunomodulatory Role for Peroxisomes in Macrophages Activated by the TLR4 Ligand Lipopolysaccharide. J. Immunol. 2017, 198, 2414–2425. [Google Scholar] [CrossRef] [Green Version]
- Di Cara, F.; Sheshachalam, A.; Braverman, N.E.; Rachubinski, R.A.; Simmonds, A.J. Peroxisome-Mediated Metabolism Is Required for Immune Response to Microbial Infection. Immunity 2017, 47, 93–106. [Google Scholar] [CrossRef]
- Facciotti, F.; Ramanjaneyulu, G.S.; Lepore, M.; Sansano, S.; Cavallari, M.; Kistowska, M.; Forss-Petter, S.; Ni, G.; Colone, A.; Singhal, A.; et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat. Immunol. 2012, 13, 474–480. [Google Scholar] [CrossRef]
- Terlecky, S.R.; Terlecky, L.J.; Giordano, C.R. Peroxisomes, oxidative stress, and inflammation. World J. Biol. Chem. 2012, 3, 93–97. [Google Scholar] [CrossRef]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar] [CrossRef]
- Su, R.J.; Jonas, B.A.; Welborn, J.; Gregg, J.P.; Chen, M. Chronic eosinophilic leukemia, NOS with t(5;12)(q31;p13)/ETV6-ACSL6 gene fusion: A novel variant of myeloid proliferative neoplasm with eosinophilia. Hum. Pathol. 2016, 5, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Geisbrecht, B.V.; Gould, S.J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J. Biol. Chem. 1999, 274, 30527–30533. [Google Scholar] [CrossRef]
- Visser, W.F.; van Roermund, C.W.; Ijlst, L.; Hellingwerf, K.J.; Waterham, H.R.; Wanders, R.J. First identification of a 2-ketoglutarate/isocitrate transport system in mammalian peroxisomes and its characterization. Biochem. Biophys. Res. Commun. 2006, 348, 1224–1231. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Hayashi, M.; Takeuchi, A.; Okamoto, T.; Kawashima, S.; Takii, T.; Hayashi, H.; Onozaki, K. Identification of a novel growth-promoting factor with a wide target cell spectrum from various tumor cells as catalase. J. Biochem. 1996, 120, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Lledias, F.; Hansberg, W. Oxidation of human catalase by singlet oxygen in myeloid leukemia cells. Photochem. Photobiol. 1999, 70, 887–892. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.D.; Parish, I.A.; Krause, D.S.; Kaech, S.M.; Shadel, G.S. Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol. Ther. 2013, 21, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Li, K.W.; Seitz, J.; Volkl, A.; Luers, G.H. Hitchhiking of Cu/Zn superoxide dismutase to peroxisomes—Evidence for a natural piggyback import mechanism in mammals. Traffic 2009, 10, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Kan, W.M. Down-regulation of superoxide dismutase 1 by PMA is involved in cell fate determination and mediated via protein kinase D2 in myeloid leukemia cells. Biochim. Biophys. Acta 2015, 1853, 2662–2675. [Google Scholar] [CrossRef] [Green Version]
- Gowing, G.; Philips, T.; Van Wijmeersch, B.; Audet, J.N.; Dewil, M.; Van Den Bosch, L.; Billiau, A.D.; Robberecht, W.; Julien, J.P. Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J. Neurosci. 2008, 28, 10234–10244. [Google Scholar] [CrossRef]
- Trompier, D.; Savary, S. X-Linked Adrenoleukodystrophy; Morgan & Claypool: Williston, VT, USA, 2013; Volume 2. [Google Scholar]
- Griffin, D.E.; Moser, H.W.; Mendoza, Q.; Moench, T.R.; O’Toole, S.; Moser, A.B. Identification of the inflammatory cells in the central nervous system of patients with adrenoleukodystrophy. Ann. Neurol. 1985, 18, 660–664. [Google Scholar] [CrossRef]
- Weber, F.D.; Wiesinger, C.; Forss-Petter, S.; Regelsberger, G.; Einwich, A.; Weber, W.H.; Kohler, W.; Stockinger, H.; Berger, J. X-linked adrenoleukodystrophy: Very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum. Mol. Genet. 2014, 23, 2542–2550. [Google Scholar] [CrossRef]
- Violante, S.; Achetib, N.; van Roermund, C.W.T.; Hagen, J.; Dodatko, T.; Vaz, F.M.; Waterham, H.R.; Chen, H.; Baes, M.; Yu, C.; et al. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019, 33, 4355–4364. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Jimenez-Sanchez, G.; Koster, J.; Denis, S.; Van Roermund, C.W.; Silva-Zolezzi, I.; Moser, A.B.; Visser, W.F.; Gulluoglu, M.; Durmaz, O.; et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 2015, 24, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Muneer, Z.; Wiesinger, C.; Voigtlander, T.; Werner, H.B.; Berger, J.; Forss-Petter, S. Abcd2 is a strong modifier of the metabolic impairments in peritoneal macrophages of abcd1-deficient mice. PLoS ONE 2014, 9, e108655. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Zierfuss, B.; Hametner, S.; Wagner, M.; Popitsch, N.; Machacek, C.; Bartolini, B.; Zlabinger, G.; Ohradanova-Repic, A.; Stockinger, H.; et al. Impaired plasticity of macrophages in X-linked adrenoleukodystrophy. Brain 2018, 141, 2329–2342. [Google Scholar] [CrossRef] [PubMed]
- Raas, Q.; Gondcaille, C.; Hamon, Y.; Leoni, V.; Caccia, C.; Ménétrier, F.; Lizard, G.; Trompier, D.; Savary, S. CRISPR/Cas9-mediated knockout of Abcd1 and Abcd2 genes in BV-2 cells: Novel microglial models for X-linked Adrenoleukodystrophy. Biochim. Biophys. Acta 2019, 1864, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Raas, Q.; Saih, F.E.; Gondcaille, C.; Trompier, D.; Hamon, Y.; Leoni, V.; Caccia, C.; Nasser, B.; Jadot, M.; Menetrier, F.; et al. A microglial cell model for acyl-CoA oxidase 1 deficiency. Biochim. Biophys. Acta 2019, 1864, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Beckers, L.; Geric, I.; Stroobants, S.; Beel, S.; Van Damme, P.; D’Hooge, R.; Baes, M. Microglia lacking a peroxisomal beta-oxidation enzyme chronically alter their inflammatory profile without evoking neuronal and behavioral deficits. J. Neuroinflamm. 2019, 16, 61. [Google Scholar] [CrossRef]
- Ishizuka, M.; Toyama, Y.; Watanabe, H.; Fujiki, Y.; Takeuchi, A.; Yamasaki, S.; Yuasa, S.; Miyazaki, M.; Nakajima, N.; Taki, S.; et al. Overexpression of human acyl-CoA thioesterase upregulates peroxisome biogenesis. Exp. Cell Res. 2004, 297, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Enayetallah, A.E.; French, R.A.; Barber, M.; Grant, D.F. Cell-specific subcellular localization of soluble epoxide hydrolase in human tissues. J. Histochem. Cytochem. 2006, 54, 329–335. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Magalhaes-Novais, S.; Bermejo-Millo, J.C.; Loureiro, R.; Mesquita, K.A.; Domingues, M.R.; Maciel, E.; Melo, T.; Baldeiras, I.; Erickson, J.R.; Holy, J.; et al. Cell quality control mechanisms maintain stemness and differentiation potential of P19 embryonic carcinoma cells. Autophagy 2019, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Copin, R.; Vitry, M.A.; Hanot Mambres, D.; Machelart, A.; De Trez, C.; Vanderwinden, J.M.; Magez, S.; Akira, S.; Ryffel, B.; Carlier, Y.; et al. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog. 2012, 8, e1002575. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Makou, P.; Finke, A.; Mielke, M. Inflammatory response of TLR4 deficient spleen macrophages (CRL 2471) to Brucella abortus S19 and an isogenic DeltamglA deletion mutant. Int. J. Med. Microbiol. 2016, 306, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Assadi, G.; Vesterlund, L.; Bonfiglio, F.; Mazzurana, L.; Cordeddu, L.; Schepis, D.; Mjosberg, J.; Ruhrmann, S.; Fabbri, A.; Vukojevic, V.; et al. Functional Analyses of the Crohn’s Disease Risk Gene LACC1. PLoS ONE 2016, 11, e0168276. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Boroviak, K.; Zhang, Q.; Assadi, G.; Kempster, S.L.; Sewell, G.W.; Saveljeva, S.; Ashcroft, J.W.; Clare, S.; Mukhopadhyay, S.; et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 2016, 17, 1046–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skon-Hegg, C.; Zhang, J.; Wu, X.; Sagolla, M.; Ota, N.; Wuster, A.; Tom, J.; Doran, E.; Ramamoorthi, N.; Caplazi, P.; et al. LACC1 Regulates TNF and IL-17 in Mouse Models of Arthritis and Inflammation. J. Immunol. 2019, 202, 183–193. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Giacoppo, S.; Bramanti, P.; Mazzon, E. NLRP3 Inflammasome Activation in a Transgenic Amyotrophic Lateral Sclerosis Model. Inflammation 2018, 41, 93–103. [Google Scholar] [CrossRef]
- Jaudszus, A.; Gruen, M.; Watzl, B.; Ness, C.; Roth, A.; Lochner, A.; Barz, D.; Gabriel, H.; Rothe, M.; Jahreis, G. Evaluation of suppressive and pro-resolving effects of EPA and DHA in human primary monocytes and T-helper cells. J. Lipid Res. 2013, 54, 923–935. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef]
- Hjorth, E.; Freund-Levi, Y. Immunomodulation of microglia by docosahexaenoic acid and eicosapentaenoic acid. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Tsao, Y.Y.; Leung, Y.M.; Su, K.P. Docosahexaenoic acid suppresses neuroinflammatory responses and induces heme oxygenase-1 expression in BV-2 microglia: Implications of antidepressant effects for omega-3 fatty acids. Neuropsychopharmacology 2010, 35, 2238–2248. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, K.C.; Jin, C.Y.; Han, M.H.; Park, C.; Lee, K.J.; Park, Y.M.; Choi, Y.H.; Kim, G.Y. Inhibitory effects of eicosapentaenoic acid on lipopolysaccharide-induced activation in BV2 microglia. Int. Immunopharmacol. 2007, 7, 222–229. [Google Scholar] [CrossRef]
- Brautigam, C.; Engelmann, B.; Reiss, D.; Reinhardt, U.; Thiery, J.; Richter, W.O.; Brosche, T. Plasmalogen phospholipids in plasma lipoproteins of normolipidemic donors and patients with hypercholesterolemia treated by LDL apheresis. Atherosclerosis 1996, 119, 77–88. [Google Scholar] [CrossRef]
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and functions. Prog. Lipid Res. 2001, 40, 199–229. [Google Scholar] [CrossRef]
- Puleston, D.J.; Villa, M.; Pearce, E.L. Ancillary Activity: Beyond Core Metabolism in Immune Cells. Cell Metab. 2017, 26, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, W.J.; Olivier, L.M.; Krisans, S.K. Central role of peroxisomes in isoprenoid biosynthesis. Prog. Lipid Res. 2002, 41, 369–391. [Google Scholar] [CrossRef]
- Jang, J.; Park, S.; Jin Hur, H.; Cho, H.J.; Hwang, I.; Pyo Kang, Y.; Im, I.; Lee, H.; Lee, E.; Yang, W.; et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun. 2016, 7, 13129. [Google Scholar] [CrossRef] [Green Version]
- Nury, T.; Zarrouk, A.; Ragot, K.; Debbabi, M.; Riedinger, J.M.; Vejux, A.; Aubourg, P.; Lizard, G. 7-Ketocholesterol is increased in the plasma of X-ALD patients and induces peroxisomal modifications in microglial cells: Potential roles of 7-ketocholesterol in the pathophysiology of X-ALD. J. Steroid Biochem. Mol. Biol. 2017, 169, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, A.; Vejux, A.; Mackrill, J.; O’Callaghan, Y.; Hammami, M.; O’Brien, N.; Lizard, G. Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res. Rev. 2014, 18, 148–162. [Google Scholar] [CrossRef]
- Oruqaj, G.; Karnati, S.; Vijayan, V.; Kotarkonda, L.K.; Boateng, E.; Zhang, W.; Ruppert, C.; Gunther, A.; Shi, W.; Baumgart-Vogt, E. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E2048–E2057. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Blumberg, B.M.; Mock, D.J.; Goodman, A.D.; Moser, A.B.; Moser, H.W.; Smith, K.D.; Powers, J.M. Potential environmental and host participants in the early white matter lesion of adreno-leukodystrophy: Morphologic evidence for CD8 cytotoxic T cells, cytolysis of oligodendrocytes, and CD1-mediated lipid antigen presentation. J. Neuropathol. Exp. Neurol. 2001, 60, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Liu, Y.; Moser, A.B.; Moser, H.W. The inflammatory myelinopathy of adreno-leukodystrophy: Cells, effector molecules, and pathogenetic implications. J. Neuropathol. Exp. Neurol. 1992, 51, 630–643. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, M.C.; Griffin, D.E.; Raymond, G.V.; Washington, C.A.; Moser, H.W.; Smith, K.D. Tumor necrosis factor-alpha and X-linked adrenoleukodystrophy. J. Neuroimmunol. 1995, 61, 161–169. [Google Scholar] [CrossRef]
- McGuinness, M.C.; Powers, J.M.; Bias, W.B.; Schmeckpeper, B.J.; Segal, A.H.; Gowda, V.C.; Wesselingh, S.L.; Berger, J.; Griffin, D.E.; Smith, K.D. Human leukocyte antigens and cytokine expression in cerebral inflammatory demyelinative lesions of X-linked adrenoleukodystrophy and multiple sclerosis. J. Neuroimmunol. 1997, 75, 174–182. [Google Scholar] [CrossRef]
- Lannuzel, A.; Aubourg, P.; Tardieu, M. Excessive production of tumour necrosis factor alpha by peripheral blood mononuclear cells in X-linked adrenoleukodystrophy. Eur. J. Paediatr. Neurol. 1998, 2, 27–32. [Google Scholar] [CrossRef]
- Di Biase, A.; Merendino, N.; Avellino, C.; Cappa, M.; Salvati, S. Th 1 cytokine production by peripheral blood mononuclear cells in X-linked adrenoleukodystrophy. J. Neurol. Sci. 2001, 182, 161–165. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Gilg, A.G.; Khan, M.; Singh, A.K.; Barbosa, E.; Singh, I. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: Implications for potential therapies. Neurobiol. Dis. 2003, 14, 425–439. [Google Scholar] [CrossRef]
- Marchetti, D.P.; Donida, B.; Jacques, C.E.; Deon, M.; Hauschild, T.C.; Koehler-Santos, P.; de Moura Coelho, D.; Coitinho, A.S.; Jardim, L.B.; Vargas, C.R. Inflammatory profile in X-linked adrenoleukodystrophy patients: Understanding disease progression. J. Cell Biochem. 2018, 119, 1223–1233. [Google Scholar] [CrossRef]
- Lund, T.C.; Stadem, P.S.; Panoskaltsis-Mortari, A.; Raymond, G.; Miller, W.P.; Tolar, J.; Orchard, P.J. Elevated cerebral spinal fluid cytokine levels in boys with cerebral adrenoleukodystrophy correlates with MRI severity. PLoS ONE 2012, 7, e32218. [Google Scholar] [CrossRef]
- Thibert, K.A.; Raymond, G.V.; Nascene, D.R.; Miller, W.P.; Tolar, J.; Orchard, P.J.; Lund, T.C. Cerebrospinal fluid matrix metalloproteinases are elevated in cerebral adrenoleukodystrophy and correlate with MRI severity and neurologic dysfunction. PLoS ONE 2012, 7, e50430. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, H.I.; Vluggens, A.; Andreoletti, P.; Ragot, K.; Mandard, S.; Kersten, S.; Waterham, H.R.; Lizard, G.; Wanders, R.J.; Reddy, J.K.; et al. The inflammatory response in acyl-CoA oxidase 1 deficiency (pseudoneonatal adrenoleukodystrophy). Endocrinology 2012, 153, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, N.; Shimada, K.; Miyazaki, T.; Kume, A.; Kitamura, Y.; Sumiyoshi, K.; Kiyanagi, T.; Iesaki, T.; Inoue, N.; Daida, H. Enhanced production of nitric oxide, reactive oxygen species, and pro-inflammatory cytokines in very long chain saturated fatty acid-accumulated macrophages. Lipids Health Dis. 2008, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Khan, M.; Singh, I. Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: Implication for X-adrenoleukodystrophy. J. Lipid Res. 2009, 50, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Singh, A.K.; Contreras, M.A. Peroxisomal dysfunction in inflammatory childhood white matter disorders: An unexpected contributor to neuropathology. J. Child Neurol. 2009, 24, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Contreras, M.A.; Gilg, A.G.; Singh, I. Oxidative imbalance in nonstimulated X-adrenoleukodystrophy-derived lymphoblasts. Dev. Neurosci. 2008, 30, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Green, S.P.; Cairns, B.; Rae, J.; Errett-Baroncini, C.; Hongo, J.A.; Erickson, R.W.; Curnutte, J.T. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 2001, 21, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, S.; Lopez-Erauskin, J.; Galino, J.; Duval, C.; Naudi, A.; Jove, M.; Kemp, S.; Villarroya, F.; Ferrer, I.; Pamplona, R.; et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet. 2008, 17, 1762–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, S.; Ruiz, M.; Guilera, C.; Hahnen, E.; Brichta, L.; Naudi, A.; Portero-Otin, M.; Dacremont, G.; Cartier, N.; Wanders, R.; et al. Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2010, 19, 2005–2014. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.M.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martinez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [Green Version]
- Galino, J.; Ruiz, M.; Fourcade, S.; Schluter, A.; Lopez-Erauskin, J.; Guilera, C.; Jove, M.; Naudi, A.; Garcia-Arumi, E.; Andreu, A.L.; et al. Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X-adrenoleukodystrophy. Antioxid. Redox Signal. 2011, 15, 2095–2107. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A.; Espinosa, L.; Fourcade, S.; Galino, J.; Lopez, E.; Ilieva, E.; Morato, L.; Asheuer, M.; Cook, T.; McLaren, A.; et al. Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy. Hum. Mol. Genet. 2012, 21, 1062–1077. [Google Scholar] [CrossRef] [PubMed]
- Leszek, J.; Barreto, G.E.; Gasiorowski, K.; Koutsouraki, E.; Avila-Rodrigues, M.; Aliev, G. Inflammatory Mechanisms and Oxidative Stress as Key Factors Responsible for Progression of Neurodegeneration: Role of Brain Innate Immune System. CNS Neurol. Disord. Drug Targets 2016, 15, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Kovacs, G.G.; Hoftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Honigschnabl, S.; Gleiss, A.; Brugger, B.; Wanders, R.; et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Rice, C.; Hares, K.; Redondo, J.; Kemp, K.; Williams, M.; Brown, A.; Scolding, N.; Wilkins, A. Reductions in neuronal peroxisomes in multiple sclerosis grey matter. Mult. Scler. 2014, 20, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Lizard, G.; Rouaud, O.; Demarquoy, J.; Cherkaoui-Malki, M.; Iuliano, L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J. Alzheimers Dis. 2012, 29, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, L.; Li, S.; Huang, J.; Chen, H.; Hou, L.; Wang, J.; Green, C.D.; Yan, Z.; Huang, X.; et al. Midlife gene expressions identify modulators of aging through dietary interventions. Proc. Natl. Acad. Sci. USA 2012, 109, E1201–E1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef] [PubMed]
- Ahlemeyer, B.; Gottwald, M.; Baumgart-Vogt, E. Deletion of a single allele of the Pex11beta gene is sufficient to cause oxidative stress, delayed differentiation and neuronal death in mouse brain. Dis. Model Mech. 2012, 5, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheckhuber, C.Q.; Erjavec, N.; Tinazli, A.; Hamann, A.; Nystrom, T.; Osiewacz, H.D. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat. Cell. Biol. 2007, 9, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.D.; Kumar, S.; van der Klei, I.J. Inhibition of peroxisome fission, but not mitochondrial fission, increases yeast chronological lifespan. Cell Cycle 2015, 14, 1698–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Auwerx, J. Calorie restriction: Is AMPK a key sensor and effector? Physiology 2011, 26, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017, 26, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, S.D.; Macdonald, S.J.; Marguerie, R.; Certa, U.; Stearns, S.C.; Goldstein, D.B.; Partridge, L. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr. Biol. 2002, 12, 712–723. [Google Scholar] [CrossRef]
- Narayan, V.; Ly, T.; Pourkarimi, E.; Murillo, A.B.; Gartner, A.; Lamond, A.I.; Kenyon, C. Deep Proteome Analysis Identifies Age-Related Processes in C. elegans. Cell Syst. 2016, 3, 144–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Mockett, R.J.; Benes, J.J.; Sohal, R.S. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J. Biol. Chem. 2003, 278, 26418–26422. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef]
- Mockett, R.J.; Bayne, A.C.; Kwong, L.K.; Orr, W.C.; Sohal, R.S. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic. Biol. Med. 2003, 34, 207–217. [Google Scholar] [CrossRef]
- Russo, M.; Di Franco, A.; Martelli, G.P. The fine structure of Cymbidium ringspot virus infections in host tissues. III. Role of peroxisomes in the genesis of multivesicular bodies. J. Ultrastruct. Res. 1983, 82, 52–63. [Google Scholar] [CrossRef]
- Uchida, T.; Suzuki, K.; Esumi, M.; Arii, M.; Oomura, M.; Shikata, T. Occurrence and ultrastructural localization of duck hepatitis B virus in the liver of ducks after experimental infection. Hepatology 1987, 7, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Reinke, P.; David, H.; Uerlings, I.; Decker, T. Pathology of hepatic peroxisomes in chronic hepatitis B and immunosuppression. Exp. Pathol. 1988, 34, 71–77. [Google Scholar] [CrossRef]
- De Craemer, D.; Bingen, A.; Langendries, M.; Martin, J.P.; Roels, F. Alterations of hepatocellular peroxisomes in viral hepatitis in the mouse. J. Hepatol. 1990, 11, 145–152. [Google Scholar] [CrossRef]
- Schaffner, F. Intralobular changes in hepatocytes and the electron microscopic mesenchymal response in acute viral hepatitis. Medicine 1966, 45, 547–552. [Google Scholar] [CrossRef]
- Tanner, L.B.; Chng, C.; Guan, X.L.; Lei, Z.; Rozen, S.G.; Wenk, M.R. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res. 2014, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sychev, Z.E.; Hu, A.; DiMaio, T.A.; Gitter, A.; Camp, N.D.; Noble, W.S.; Wolf-Yadlin, A.; Lagunoff, M. Integrated systems biology analysis of KSHV latent infection reveals viral induction and reliance on peroxisome mediated lipid metabolism. PLoS Pathog. 2017, 13, e1006256. [Google Scholar] [CrossRef] [PubMed]
- Jean Beltran, P.M.; Cook, K.C.; Hashimoto, Y.; Galitzine, C.; Murray, L.A.; Vitek, O.; Cristea, I.M. Infection-Induced Peroxisome Biogenesis Is a Metabolic Strategy for Herpesvirus Replication. Cell Host Microbe 2018, 24, 526–541.e7. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.B.; Choi, Y.; Harhaj, E.W. Peroxisomes support human herpesvirus 8 latency by stabilizing the viral oncogenic protein vFLIP via the MAVS-TRAF complex. PLoS Pathog. 2018, 14, e1007058. [Google Scholar] [CrossRef]
- Zhou, M.T.; Qin, Y.; Li, M.; Chen, C.; Chen, X.; Shu, H.B.; Guo, L. Quantitative Proteomics Reveals the Roles of Peroxisome-associated Proteins in Antiviral Innate Immune Responses. Mol. Cell. Proteom. 2015, 14, 2535–2549. [Google Scholar] [CrossRef]
- Miyake, K.; Shibata, T.; Ohto, U.; Shimizu, T.; Saitoh, S.I.; Fukui, R.; Murakami, Y. Mechanisms controlling nucleic acid-sensing Toll-like receptors. Int. Immunol. 2018, 30, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, C.; Beachboard, D.C.; Horner, S.M. Methods to Visualize MAVS Subcellular Localization. Methods Mol. Biol. 2017, 1656, 131–142. [Google Scholar] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, B.; Li, Y.; Zhu, L.; Li, P.; Zou, F.; Bin, L. The Solute Carrier Transporter SLC15A3 Participates in Antiviral Innate Immune Responses against Herpes Simplex Virus-1. J. Immunol. Res. 2018, 2018, 5214187. [Google Scholar] [CrossRef]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus Infection Impairs Peroxisome Biogenesis and Early Antiviral Signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Asahchop, E.L.; Branton, W.G.; Gelman, B.B.; Power, C.; Hobman, T.C. MicroRNAs upregulated during HIV infection target peroxisome biogenesis factors: Implications for virus biology, disease mechanisms and neuropathology. PLoS Pathog. 2017, 13, e1006360. [Google Scholar] [CrossRef]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Magalhaes, A.C.; Camoes, F.; Gouveia, A.; Vieira, M.; Kagan, J.C.; Ribeiro, D. Hepatitis C virus NS3-4A inhibits the peroxisomal MAVS-dependent antiviral signalling response. J. Cell. Mol. Med. 2016, 20, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kang, J.A.; Han, M.H.; Chung, K.H.; Lee, C.R.; Song, W.K.; Jun, Y.; Park, S.G. Peroxisome-localized hepatitis Bx protein increases the invasion property of hepatocellular carcinoma cells. Arch. Virol. 2014, 159, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, M.; Whelband, M.; Mohorianu, I.; Powell, P.P. The pestivirus N terminal protease N(pro) redistributes to mitochondria and peroxisomes suggesting new sites for regulation of IRF3 by N(pro.). PLoS ONE 2014, 9, e88838. [Google Scholar] [CrossRef]
- Cohen, G.B.; Rangan, V.S.; Chen, B.K.; Smith, S.; Baltimore, D. The human thioesterase II protein binds to a site on HIV-1 Nef critical for CD4 down-regulation. J. Biol. Chem. 2000, 275, 23097–23105. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Margottin, F.; Le Gall, S.; Schwartz, O.; Selig, L.; Benarous, R.; Benichou, S. Binding of HIV-1 Nef to a novel thioesterase enzyme correlates with Nef-mediated CD4 down-regulation. J. Biol. Chem. 1997, 272, 13779–13785. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Shiratori, T.; Shoji, H.; Miyatake, S.; Okazaki, Y.; Ikuta, K.; Sato, T.; Saito, T. A novel acyl-CoA thioesterase enhances its enzymatic activity by direct binding with HIV Nef. Biochem. Biophys. Res. Commun. 1997, 238, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Grinstein, S.; Roth, Z. Diversity and Versatility of Phagocytosis: Roles in Innate Immunity, Tissue Remodeling, and Homeostasis. Front. Cell. Infect. Microbiol. 2017, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Bergner, C.G.; van der Meer, F.; Winkler, A.; Wrzos, C.; Turkmen, M.; Valizada, E.; Fitzner, D.; Hametner, S.; Hartmann, C.; Pfeifenbring, S.; et al. Microglia damage precedes major myelin breakdown in X-linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia 2019, 67, 1196–1209. [Google Scholar] [CrossRef]
- Gong, Y.; Sasidharan, N.; Laheji, F.; Frosch, M.; Musolino, P.; Tanzi, R.; Kim, D.Y.; Biffi, A.; El Khoury, J.; Eichler, F. Microglial dysfunction as a key pathological change in adrenomyeloneuropathy. Ann. Neurol. 2017, 82, 813–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.R.; Cauvi, D.M.; Rivera, I.; Hawisher, D.; De Maio, A. Changes in macrophage function modulated by the lipid environment. Innate Immun. 2016, 22, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.B.; Liao, Y.C.; Qi, W.; Xie, C.; Du, X.; Wang, J.; Yang, H.; Miao, H.H.; Li, B.L.; Song, B.L. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 2015, 161, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjorth, E.; Zhu, M.; Toro, V.C.; Vedin, I.; Palmblad, J.; Cederholm, T.; Freund-Levi, Y.; Faxen-Irving, G.; Wahlund, L.O.; Basun, H.; et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-beta42 by human microglia and decrease inflammatory markers. J. Alzheimers Dis. 2013, 35, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Bond, J.A.; Harvey, D.J.; Gordon, S.; Newsholme, E.A. Uptake and incorporation of saturated and unsaturated fatty acids into macrophage lipids and their effect upon macrophage adhesion and phagocytosis. Biochem. J. 1990, 269, 807–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, S.; Fuhrmann, H.; Schumann, J. Unsaturated fatty acids promote the phagocytosis of P. aeruginosa and R. equi by RAW264.7 macrophages. Curr. Microbiol. 2012, 65, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, D.S.; Hansen, J.B.; Grimsgaard, S.; Bonaa, K.H.; Kierulf, P.; Nordoy, A. The effect of highly purified eicosapentaenoic and docosahexaenoic acids on monocyte phagocytosis in man. Lipids 1997, 32, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Gorjao, R.; Verlengia, R.; Lima, T.M.; Soriano, F.G.; Boaventura, M.F.; Kanunfre, C.C.; Peres, C.M.; Sampaio, S.C.; Otton, R.; Folador, A.; et al. Effect of docosahexaenoic acid-rich fish oil supplementation on human leukocyte function. Clin. Nutr. 2006, 25, 923–938. [Google Scholar] [CrossRef]
- Pisani, L.F.; Lecchi, C.; Invernizzi, G.; Sartorelli, P.; Savoini, G.; Ceciliani, F. In vitro modulatory effect of omega-3 polyunsaturated fatty acid (EPA and DHA) on phagocytosis and ROS production of goat neutrophils. Vet. Immunol. Immunopathol. 2009, 131, 79–85. [Google Scholar] [CrossRef]
- Lecchi, C.; Invernizzi, G.; Agazzi, A.; Ferroni, M.; Pisani, L.F.; Savoini, G.; Ceciliani, F. In vitro modulation of caprine monocyte immune functions by omega-3 polyunsaturated fatty acids. Vet. J. 2011, 189, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Majai, G.; Sarang, Z.; Csomos, K.; Zahuczky, G.; Fesus, L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur. J. Immunol. 2007, 37, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Sannes, P.L.; Spicer, S.S. Peroxisomes of rat peritoneal macrophages during phagocytosis. Am. J. Pathol. 1979, 95, 281–294. [Google Scholar] [PubMed]
- Dahabieh, M.S.; Di Pietro, E.; Jangal, M.; Goncalves, C.; Witcher, M.; Braverman, N.E.; Del Rincon, S.V. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 103–121. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A-Proteins with a bona fide (strict or partial) peroxisomal localization | |||||
| Protein | Function and Diseases | Immune Process | Cell or Tissue Type | Organism * | Ref |
| ABCD1 | Fatty acid oxidation ABC transporter X-ALD (MIM 300100) | Phagocytosis? Microglial homeostasis? Inflammation | Microglia (BV-2) | m | [56] |
| CD14+ monocytes | h | [55] | |||
| Macrophages | h | [55] | |||
| PBMCs | h | [51] | |||
| ABCD2 | Fatty acid oxidation ABC transporter | Phagocytosis? Microglial homeostasis? Inflammation? ABCD1 compensation | Microglia (BV-2) | m | [56] |
| Macrophages (peritoneal) | m | [54] | |||
| PBMCs | h | [51] | |||
| ABCD3 | Fatty acid oxidation ABC transporter CBAS5 (MIM 616278) | ABCD1 compensation | PBMCs | h | [51] |
| ACOT8 | Fatty acid oxidation Lipid metabolism | Antiviral response Peroxisome proliferation | T lymphoma (Jurkat E6.1) | h | [59] |
| ACOX1 | Fatty acid oxidation ACOX1 Def. (MIM 264470) | Phagocytosis? Microglial homeostasis? Inflammation | Microglia (BV-2) | m | [57] |
| ACSL6 | Lipid metabolism | Cell proliferation, Leukemogenesis | Bone marrow | h | [35] |
| Myeloid leukemia cells | h | [34] | |||
| CAT | Antioxidant system Acatalasemia (MIM 614097) | Cell proliferation Peroxisome proliferation | Lymph nodes | h | [60] |
| T lymphoma (Jurkat E6.1) | h | [59] | |||
| Myeloid leukemia cells U937 | h | [44] | |||
| T hybridoma (2B4), B lymphoma A20, mast cell P815 | m | [59] | |||
| Microglia (BV-2) | m | [57] | |||
| Macrophages | d | [31] | |||
| DNM1L | Peroxisome biogenesis Encephalopathy (MIM 614388) | Mitochondrial and peroxisomal fission | Microglia progenitor cells | h | [61,62] |
| GNPAT | Ether lipid synthesis RCDP2 (MIM 222765) | Cell proliferation Maturation of iNKT cells | Thymocytes | m | [32] |
| MAVS | RIG-I-like receptor (RLR) adaptor | Antiviral response | Macrophages | m | [63] |
| MFP2 | Fatty acid oxidation MFP2 Def. (MIM 261515) | Inflammation | Microglia (brain) | m | [58] |
| Macrophages (Raw) | m | [30] | |||
| NOS2 | Antioxidant system | Brucellosis pathogenesis Inflammation | Dendritic cells and monocytes | m | [64] |
| CRL 2471 spleen macrophages | m | [65] | |||
| PEX5 | Peroxisome biogenesis PBD2A (MIM214110), PBDBB (MIM 202370), RCDP5 (MIM 616716) | Inflammation NF-κB regulation | Macrophages | m | [31] |
| d | [30,31] | ||||
| PEX7 | Peroxisome biogenesis RCDP1 (MIM 215100) | Inflammation NF-κB regulation | Macrophages | m | [31] |
| d | [31] | ||||
| PEX14 | Peroxisome biogenesis PBD13A (MIM 614887) | Inflammation NF-κB regulation | Macrophages (Raw, primary alveolar, primary peritoneal) | m | [30] |
| B-Proteins with an ambiguous peroxisomal localization | |||||
| Protein | Function and Diseases | Immune Process | Cell or Tissue Type | Organism * | Ref |
| FAMIN LACC1 | Fatty acid oxidation | Inflammation | Neutrophils, monocytes/macrophages, dendritic cells | h | [66,67] |
| Macrophages (differentiated THP-1) | h | [66,67] | |||
| Lymph nodes, spleen | h | [66,67] | |||
| Macrophages, dendritic cells, neutrophils | m | [68] | |||
| IDH1 | Regeneration of NADPH Lipid metabolism | Hematopoietic differentiation Acute myelogenous leukemia | CD34+ bone marrow cells | h | [39] |
| Bone marrow mononuclear cells | h | [40] | |||
| Macrophages (THP-1, Kasumi-1, KG-1, OCI-AML3) | h | [41] | |||
| SOD1 | Antioxidant system | Neuroinflammation Cell proliferation | Brain | r | [69] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bülow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in Immune Response and Inflammation. Int. J. Mol. Sci. 2019, 20, 3877. https://doi.org/10.3390/ijms20163877
Di Cara F, Andreoletti P, Trompier D, Vejux A, Bülow MH, Sellin J, Lizard G, Cherkaoui-Malki M, Savary S. Peroxisomes in Immune Response and Inflammation. International Journal of Molecular Sciences. 2019; 20(16):3877. https://doi.org/10.3390/ijms20163877
Chicago/Turabian StyleDi Cara, Francesca, Pierre Andreoletti, Doriane Trompier, Anne Vejux, Margret H. Bülow, Julia Sellin, Gérard Lizard, Mustapha Cherkaoui-Malki, and Stéphane Savary. 2019. "Peroxisomes in Immune Response and Inflammation" International Journal of Molecular Sciences 20, no. 16: 3877. https://doi.org/10.3390/ijms20163877