
Molecular Mechanism of Organic Crystal Nucleation: A Perspective of Solution Chemistry and Polymorphism


1
School of Chemical Engineering and Technology, State Key Laboratory of Chemical Engineering, Tianjin University, Tianjin 300072, China
2
The Co-Innovation Center of Chemistry and Chemical Engineering of Tianjin, Tianjin 300072, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Crystals 2022, 12(7), 980; https://doi.org/10.3390/cryst12070980
Submission received: 13 June 2022
/
Revised: 9 July 2022
/
Accepted: 11 July 2022
/
Published: 14 July 2022
(This article belongs to the Special Issue Pharmaceutical Crystal and Process Engineering)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Crystal nucleation determining the formation and assembly pathway of first organic materials is the central science of various scientific disciplines such as chemical, geochemical, biological, and synthetic materials. However, our current understanding of the molecular mechanisms of nucleation remains limited. Over the past decades, the advancements of new experimental and computational techniques have renewed numerous interests in detailed molecular mechanisms of crystal nucleation, especially structure evolution and solution chemistry. These efforts bifurcate into two categories: (modified) classical nucleation theory (CNT) and non-classical nucleation mechanisms. In this review, we briefly introduce the two nucleation mechanisms and summarize current molecular understandings of crystal nucleation that are specifically applied in polymorphic crystallization systems of small organic molecules. Many important aspects of crystal nucleation including molecular association, solvation, aromatic interactions, and hierarchy in intermolecular interactions were examined and discussed for a series of organic molecular systems. The new understandings relating to molecular self-assembly in nucleating systems have suggested more complex multiple nucleation pathways that are associated with the formation and evolution of molecular aggregates in solution.
1. Introduction
Crystallization is the central topic in various disciplines such as chemical, geochemical, biological, and synthetic materials [1]. Nature utilizes crystallization in an elegant way to achieve a remarkable diversity of shapes, patterns, compositions, and functions of the arising crystalline materials from snowflakes, rocks to the bone, and teeth of ocean organisms [2,3,4]. From a chemical and synthetic material perspective, chemical and/or physical purity is one of the key attributes wherein crystallization is a well-known approach of separation and purification and has been widely used in pharmaceutical, food, dyes, and fine chemical industries [5,6,7,8]. As one of the oldest technologies, crystallization had been utilized to prepare salts from salt lakes or seawater, dating back to as early as the Ancient Yellow Emperor period (about 2700 B.C.) in China and thousands of years ago all over the world. However, despite a long history, the theory of the development of crystallization is rather slow, and significant progress was made until the late-1800s when Gibbs established the thermodynamic underpinnings of phase transition [9,10].
Crystal nucleation, the first and key step of crystallization from solution, is a typical phase transition process from liquid-phase precursors [11,12,13,14,15] creating an enormous diversity of crystalline materials. It dictates the process of molecular assembly and plays a decisive role in many properties of materials including particle shape, size distribution, purity, chirality, etc. [16,17,18]. Mechanistic understandings and the precise control of nucleation have received lots of attention in crystal engineering, process engineering, and material chemistry [16]. This is evidenced by a number of key reviews over the past decades [13,19]. For example, Kashchiev et al. and Davey et al. reviewed the classical nucleation theory (CNT) that applies to systems of small organic molecules with an emphasis on the molecular interpretation of nucleation kinetics. Myerson et al. and Vekilov [20] reviewed the evidence of the two-step nucleation mechanism where Vekilov detailed a two-step sequence scenario in which structure order is preceded by the separation of a dense, disordered liquid phase in protein systems. Gebauer and Cölfen differentiated the nature of clusters at different nucleation stages and reviewed the evidence of pre-nucleation clusters in inorganic systems [21,22]. Anwar et al. [23] and Michaelides et al. [24] reviewed computational approaches used to elucidate the molecular nature of nucleation and pointed to challenges in the simulation accuracy of interatomic potentials and enhanced sampling methods. Sleutel et al. reviewed the nucleation of protein crystals and summarized different protein nucleation models and their limitations [25].
Thanks to the advancements in analytical techniques, measurement methods, and computational simulations over the past decades [13,26,27,28], many mechanisms of crystal nucleation have been proposed which are generally classified into two categories: classical nucleation theory (CNT) versus non-classical nucleation mechanisms [13]. CNT holds that density fluctuations are concomitant with the development of crystalline order. In other words, a crystal nucleus has an identical structure to its bulk crystal. However, experimental observations and computational simulations suggest that crystalline order is preceded by fluctuations in density. On the other hand, stable associates or clusters were observed in some inorganic systems. These results invoked non-classical nucleation mechanisms such as two-step nucleation, pre-nucleation clusters, and multistep nucleation [27,29,30,31].
Moreover, the application of state-of-art modern analytic techniques such as solution spectroscopy, neutron scattering, and high-resolution electron microscopy, has spurred significant advancement in the understanding of solution chemistry and (non)-classical nucleation pathway [32,33,34]. The structure, intermolecular interactions, and assembly mode of solution associates are revealed by collectively using ATR-FTIR, 1H/13C-NMR, and advanced DOSY or NOESY NMR spectroscopy in a number of small organic molecules [35,36,37]. The dynamic evolution of solution aggregates of glycine toward crystal nucleation is probed by synchrotron-based small-angle X-ray spectroscopy [38]. The application of high-resolution cryo-TE [31,39,40,41,42] and in situ AFM [43,44] also provides detailed insights into the evolution of (non)-classical nucleation pathways. Computational simulations have achieved a remarkable computational capability and developed better sampling methods that are able to capture the rare event of nucleation at the atomic level [15].
Given these advances, the purpose of our review is to summarize the recent key advancements in the molecular interpretation of the mechanistic understanding of crystal nucleation which leads to the postulation and concept of multiple nucleation pathways for organic molecular systems, and to look into the remained open questions for future work. The review is organized as follows: first, we briefly introduce the recent development in classical nucleation theory at the molecular level and non-classical nucleation mechanisms; then we summarize the recent research progress on the molecular interpretations of crystal nucleation in organic polymorphic systems with a focus on an illustration of the relationship between solution associates or aggregates and the resultant crystal structure and hierarchy of intermolecular interactions; last, we look to future work and raise some unsolved questions about crystal nucleation, which hopefully will inspire researchers devoted to this intriguing topic of crystal nucleation.
2. Classical Nucleation Theory (CNT)
2.1. Thermodynamics
CNT was originally derived from the pioneering work of Fahrenheit on the supercooling of water in the early 1700s. It was endowed with thermodynamic underpinnings by Gibbs in the late 1800s on the studies of droplet formation on a supersaturated vapor. In the early 1900s, Volmer and Weber, and others formulated the kinetic aspects of CNT for vapor condensation; subsequently, they were extended by Turnbull and Fisher in the 1950s to address the cases of nucleation in condensed phases [45].
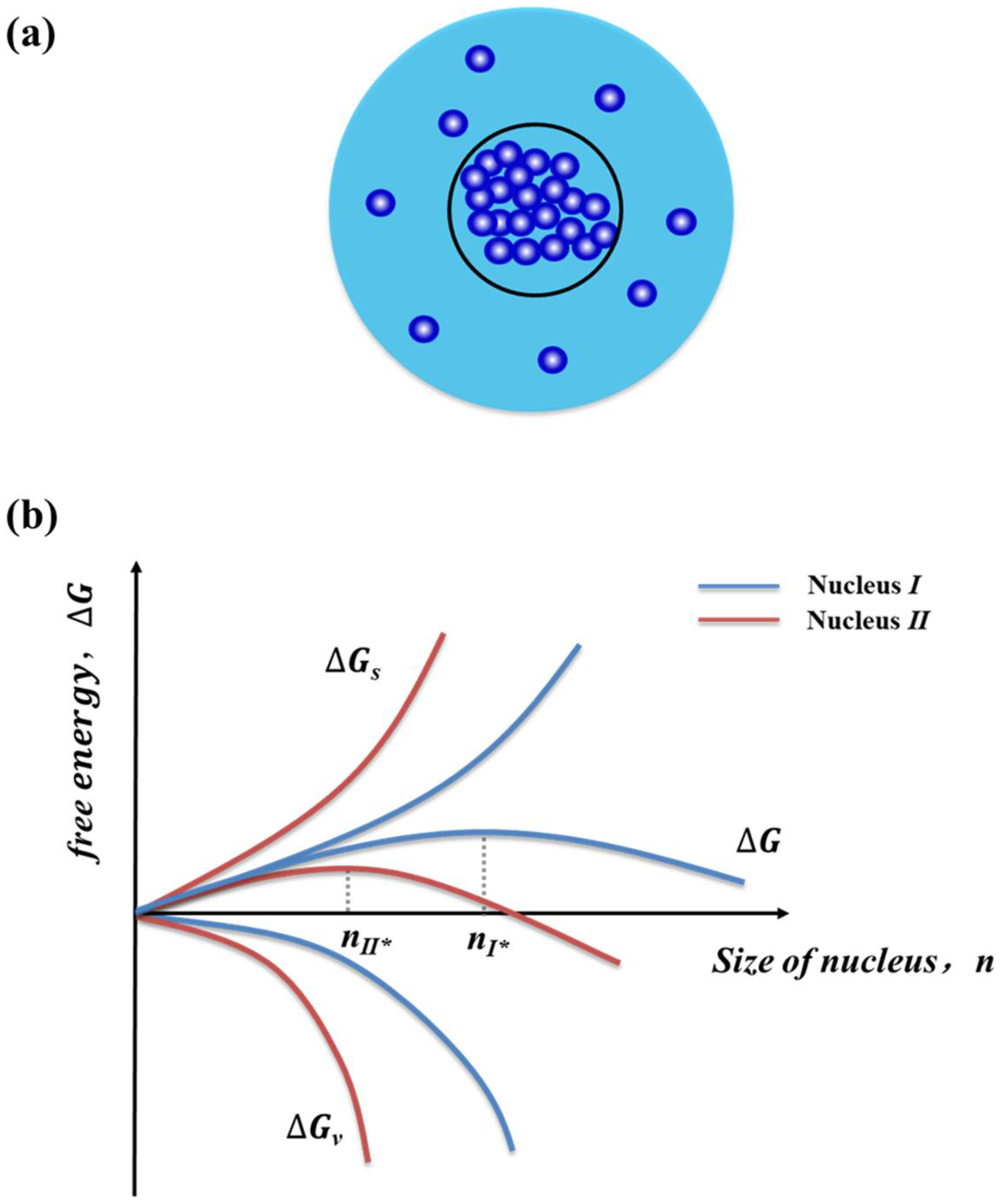
Nucleation is the first step of the phase transition that occurs by local density fluctuations of the old phase from a supersaturated state [11]. The path by which the phase transition takes place is often described by the cluster approach. A cluster of a certain number n of molecules (or building units) may be formed rarely because of a free-energy barrier presented in the course of cluster formation (Figure 1a). The barrier results from the competition in the growing clusters between the gain in bulk free energy (∆Gv = −n∆μ) due to the addition of a molecule and the increasing surface free energy (∆Gs = aγ) via the creation of the additional crystal-liquid interface with specific interfacial free energy, γ, and a surface area, a. The total free energy is thus given by
where kB is the Boltzmann constant, T is the absolute temperature, S is the solution supersaturation ratio, c is the actual concentration, c0 is the equilibrium (saturated) concentration, A is a shape factor, and v0 is the molecular volume in the crystalline phase.
When the size of a cluster is small, the contribution of the surface free energy will be dominant over the bulk free energy, and the cluster will tend to dissolve. However, the bulk free energy will become dominant for the clusters with relatively large sizes. Therefore, a critical size of cluster (n*, also named as nucleus) exists, beyond which the cluster probably survives from dissolution and becomes a nucleus growing further (Figure 1b) [46]. The critical size of a nucleus and the critical nucleation barrier (∆Gc) were given by
The size of a critical nucleus is dependent upon the strength of free energy barrier, which is influenced by crystallization conditions such as supersaturation, solvent, and temperature [13]. Higher levels of solution supersaturations (i.e., greater driven force) leads to the lower free energy barrier and thus the lower size of the critical nucleus. The critical clues and the size-dependence on the thermodynamic driving force have been recently demonstrated experimentally [47].
2.2. The Kinetics of Crystal Nucleation
Resembling the kinetic theory of reaction, the steady-state rate of nucleation (J), which is defined as the number of nuclei per unit time per unit volume, is expressed in the form of the Arrhenius rate equation:
where A is the pre-exponential factor. Equation (6) can be rewritten as
Here B represents the thermodynamic parameter. Equation (8) is valid for homogenous nucleation from the solution. In the case of heterogeneous nucleation, the interfacial energy γ should be replaced by an effective interfacial energy γHEN = φγ, where the activity factor of template substrate, φ, is in the range of 0 to 1, dependent upon the wetting ability of a cluster with the substrate surface. Note that in the case of homogeneous nucleation, the interfacial free energy represents an average over different crystal planes. More discussions on heterogeneous nucleation can be found in a book by Kashchiev [11].
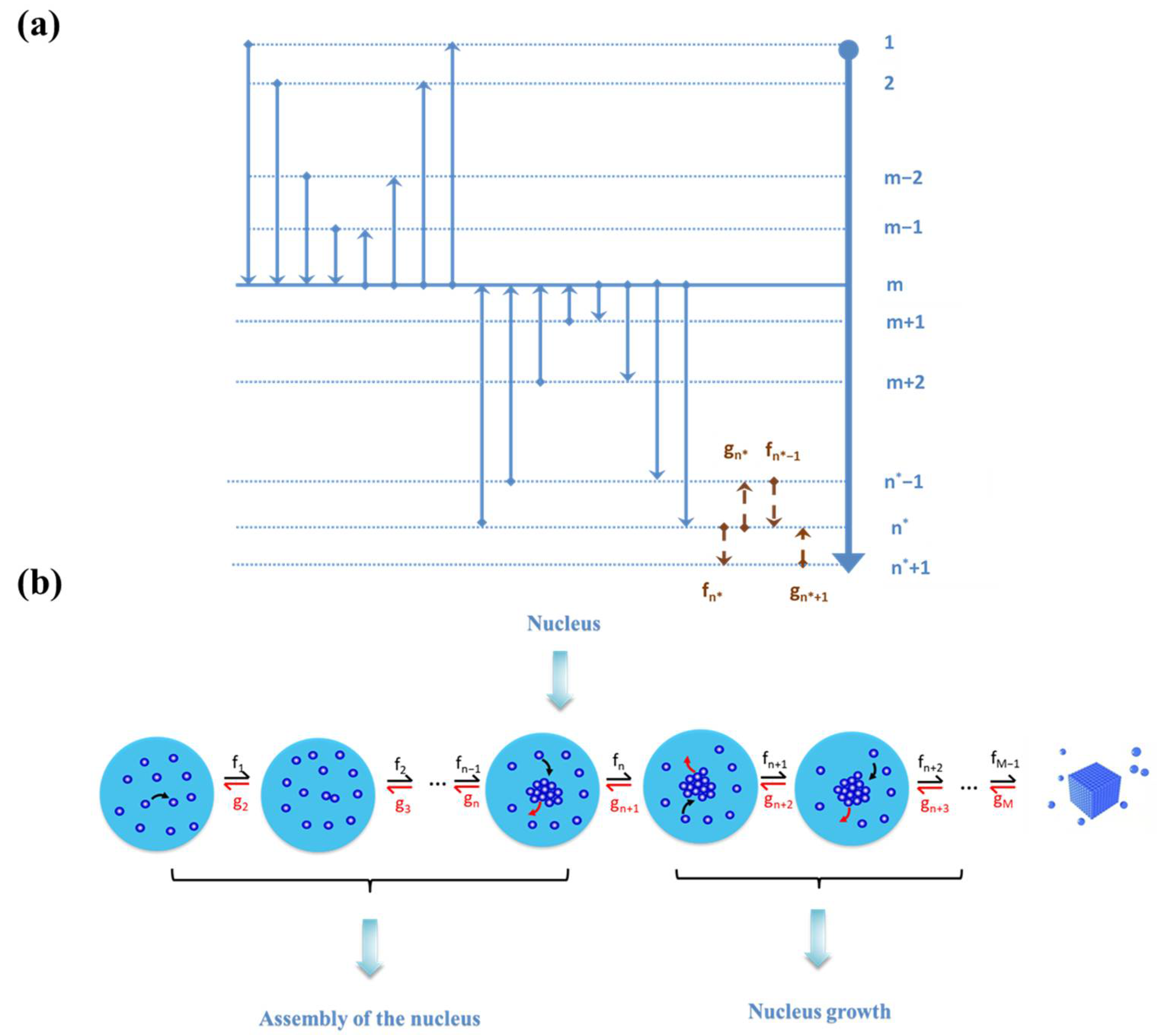
The pre-exponential factor A in Equation (6) is associated with the molecular kinetics of the nucleation process. In the framework of the cluster approach, nucleation is assumed to be a consecutive series of attachments and detachments to form differently sized clusters of the nucleating phase in the supersaturated solution (Figure 2a). The model assumes the presence of clusters of m (=2, 3, …) molecules in the old phase and transformations of m-sized clusters into n-sized ones via time-dependent frequencies fmn(t) (s−1) [11]. It is also possible that the equally sized clusters have different shapes, but the clusters of a given size have only one shape and were generally postulated for model simplification. Note that such an assumption leads to the size being the sole parameter to describe clusters and the crystal nuclei thereof. Moreover, a change in the cluster size may occur by attachment and detachment of monomers, dimers, trimers, or even higher aggregates, but a sequence of bimolecular addition (that is each addition by monomers) was often considered to be “more likely”. These assumptions lead to the well-known, simplified Szilard–Farkas model, which describes the nucleation process as a successive attachment and detachment of monomers to and from clusters of various sizes (Figure 2b).
The nucleation kinetics is controlled by the frequencies fn* and gn* of monomer attachment and detachment from an n*-sized cluster, respectively. In a stationary state of the nucleation process with a constant concentration Xn of n-size clusters, the nucleation rate was defined as the difference between the transformation frequency fn*Xn* of the nuclei (n*) into the smallest super-nuclei (n* + 1) and detachment frequency gn*+1Xn*+1 of the super-nuclei n* + 1 into the nuclei n*:
With f* ≡ fn*, X* ≡ Xn*, Equation (9) is rearranged into a familiar form
where z is the Zeldovich factor accounting for the use of C* instead of the actual nucleus concentration X* and for those clusters larger than nuclei but eventually decay rather than grow into macroscopic crystals. C* = C0exp(−∆Gc/kBT) is the equilibrium concentration of crystal nuclei. C0 is the concentration of nucleation sites in the system and is assumed equal to 1/v0 for homogenous nucleation (v0 is the volume of a single solute molecule).
The nucleation rate was thus determined by not only the nucleation barrier ∆Gc and the concentration C0 of nucleation sites but also the attachment frequency f* of monomers to the nucleus. According to Equations (7), (11), and (12), the pre-exponential factor zf*C0 is related to molecular attachment events of monomers to the critical nucleus. The kinetic attachment over the course of nucleation could be controlled either by the volume diffusion process of monomers in solution towards the nucleus or by the interfacial transfer of monomers across the interface of the crystal nucleus and the surrounding solution.
When volume diffusion was the rate-determining step in monomer attachment to the nucleus, f* = j*S* wherein S* is the surface area of a nucleus and j* is the diffusion flux of monomers to the nucleus surface. By the assumption of a spherical shape, the radius of a nucleus:
The flux j* = DC/r* and f* is given by
where D is the diffusion coefficient of monomers and C is the concentration of monomers in the bulk solution.
If the event of molecular attachment is controlled by interface transfer, the monomers are in immediate contact with the nucleus but need to make a random jump over a distance d0 ≈ (60/π)1/3 before joining into the nucleus. Assuming that such a jump is proportional to D and the sticking coefficient λ of monomers, j* = DC/d0 and S* = 4πr*2, and f* is given by
The aforementioned molecular interpretations of nucleation kinetics appear plausible and were used to qualitatively explain crystal nucleation phenomena in condensed phases. In some cases, CNT does provide a reasonable prediction on nucleation rate, e.g., the homogeneous nucleation of water droplets in vapors [48], but it fails in many other cases wherein the predicted nucleation rate often displays a few orders of magnitude derivation from measurements [49]. Even for simple mono-component nucleation in vapor condensation like methanol, the measured nucleation rate is 10−10 slower than the predicted one [50]. Several major assumptions of CNT are responsible for these discrepancies: (1) The molecular arrangement of a crystal nucleus is identical to the bulk crystalline phase, and no surface free energy or interface tension difference between the two (the so-called capillarity approximation). This assumption, however, appears incompatible with the Gibbs–Thomson effect in which the curved step displays higher surface tension than that of the straight one. In addition, the surface tension of a crystal nucleus can be also less than that at a flat interface [51,52], which is valid at least in isobaric supercooling [53,54]. (2) The evolution of crystalline order and clusters’ density occurs simultaneously in crystal nucleation, and growth and dissipation of clusters proceed respectively via the attachment and detachment of monomers (i.e., Szilard–Farkas model). However, it is possible that some organic systems displaying significant molecular association or aggregation form clusters via dimer or oligomers, not only monomers [13,38,55]. Further, the large stable ionic aggregates or clusters were also reported in solution prior to nucleation in inorganic systems [56,57]. (3) The steady-state nucleation process is assumed, and the stationary distribution of clusters is established instantaneously upon reaching the supersaturation state. In addition, the overestimated rate of crystal nucleation was often attributed to heterogeneous nucleation through foreign particles, which, nevertheless, are not well defined in both structure and concentration. Besides, recent studies have also shown that the discrepancy at high metastability is due to the nucleation times measured in not-fully relaxed liquids [58,59,60].
3. Non-Classical Nucleation Mechanisms
Over the past two decades, more and more experimental and simulation evidence [13,25,40,61,62] have demonstrated that the intermediate precursors such as dense liquid clusters/phase, amorphous phase, and soluble oligomers or aggregates [63,64] play an important role in determining the kinetics and pathway of crystal nucleation. These unconventional observations lead to the development of non-classical nucleation mechanisms. The two-step nucleation mechanism and pre-nucleation cluster (PNC) pathway may be the two representatives of these proposed mechanisms. They are different from CNT in regard to the evolution of the reaction coordinate (the reaction process from molecules, clusters to crystal) of crystal nucleation. Both mechanisms agree with the formation of intermediate precursors prior to crystal nucleation, but the precursors are suggested to be metastable dense liquid clusters or stable dense liquid phase in the two-step mechanism [65] and are considered to be stable solute clusters in the PNC pathway [66]. It is noted that the solute clusters claimed in the PNC pathway are distinct in structure and thermodynamics from the metastable clusters suggested in CNT [67].
3.1. Two-Step Nucleation Mechanism
The two-step nucleation mechanism holds that the fluctuations in density is prior to the development of the crystalline order of a critical nucleus, leading to the formation of intermediate dense liquid clusters or liquid phase, which is essentially different from CNTs that advocate the simultaneous evolution of density and crystalline orders [13] (Figure 3a). The scenario was firstly supported in computational simulations, for example by ten Wolde and Frenkel [68], using the Monte Carlo simulation technique in a Lennard–Jones colloidal system. Large density fluctuations were observed around the critical temperature where a highly disordered liquid droplet was formed prior to the development of a crystalline nucleus inside the droplet. Later on, Talanquer [69] applied the density functional theory (DFT) to the study of the homogenous nucleation of colloid and globular proteins in solution. They found that disordered droplet polymers appeared first and then nucleated into crystals, indicating that the first step towards the formation of a critical long-range ordered nucleus is the formation of a disordered liquid-like structure near the critical temperature point.
Experimental evidence to support the two-step mechanism was initially observed directly in colloidal systems [70] but was predominately reported for protein crystallization [71,72,73]. Numerous studies on protein nucleation by dynamic light and/or small-angle scattering techniques showed the first formation of fractal or droplet-like aggregates that progressively evolve into compact crystal structures [74,75,76]. For example, the metastable mesophase (MIP) of proteins was identified and its role on the evolution of crystal nucleation was revealed through the combined use of time-resolved in situ small-angle X-ray scattering (SAXS) and an optical microscope [77]. Others understood the phenomena of the nucleation of proteins through the direct measurements of nucleation kinetics with delicate experimental design [78]. One of the most prominent studies presented by Vekilov and co-workers [79] is the study of nucleation kinetics of lysozyme crystals. The nucleation rate of a lysozyme protein was measured by directly counting the number of crystals per unit volume within a certain time period, and the reproducible statistical characteristics of the random nucleation process were captured by more than 400 trials under the identical conditions. The measured kinetic dependence of protein nucleation, however, shows several unusual features that do not comply with the predictions of CNT. The results demonstrate that the nucleation of lysozyme crystals proceeds in two steps: the formation of a dense liquid droplet, followed by the nucleation of a periodic crystal within the droplet [79]. It was also concluded that the structure fluctuations to become crystalline nuclei do not require large density fluctuations or long-lifetime droplets meaning that the dense liquid is either metastable clusters or a stable phase existing below the liquid–liquid separation line (Figure 3b). More recently, it was shown that the structure development of dense liquid precursors into an ordered nucleus determines the rate of crystal nucleation [20]. However, the nucleation of proteins does not have to be a two-step process. Van Driesch et al. observed the direct self-assembly of glucose isomerase protein molecules into polyhedral nanocrystals with surprising smooth surfaces and sharp vertices, indicating a one-step nucleation mechanism [80]. The more detailed theoretical developments of the two-step nucleation mechanism can be found here [81,82].
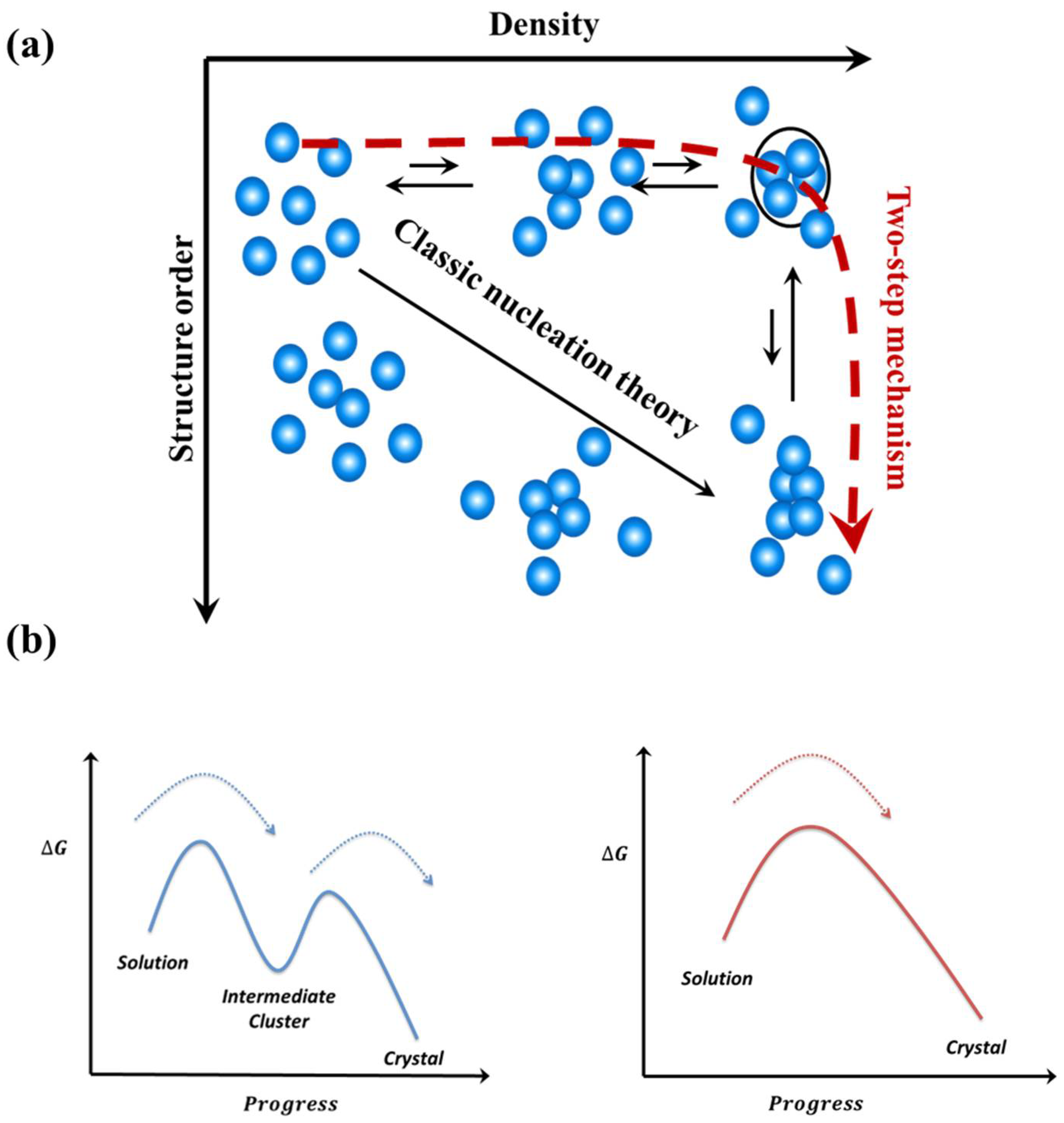
Figure 3.
The two-step nucleation mechanism: (a) Comparison of nucleation pathways of the two-step mechanism with CNT. Reprinted/adapted with permission from [72]. Copyright 2017, copyright Vekilov, P.G. et al. Copyright©2004, American Chemical Society. (b) Comparison of free energy along the pathway of crystal nucleation followed the two-step nucleation mechanism (left) and CNT (right).
Figure 3.
The two-step nucleation mechanism: (a) Comparison of nucleation pathways of the two-step mechanism with CNT. Reprinted/adapted with permission from [72]. Copyright 2017, copyright Vekilov, P.G. et al. Copyright©2004, American Chemical Society. (b) Comparison of free energy along the pathway of crystal nucleation followed the two-step nucleation mechanism (left) and CNT (right).

The two-step nucleation mechanism may also apply to small organic molecules. An early molecular dynamic simulation on acetic acid solutes in the CCl4 solvent system shows the formation of a liquid-like solute micelle, which was suggested to be the first step of crystal nucleation [83]. Experimentally, the nucleation of glycine was long-standing considered to follow the two-step mechanism on the basis of an intriguing experimental phenomenon, named non-photochemical laser-induced nucleation (NPLIN) first observed by Myerson and co-workers [84]. It was found depending on the polarization state of the laser. The α form of glycine is crystallized from an aqueous solution with circular light, whereas the γ form is obtained when linear light is introduced. The results were explained by the orientation alignment capability of pre-existed glycine clusters in solution by linearly or circularly polarized light. Further support of glycine followed the two-step nucleation mechanism was uncovered by the small-angle X-ray scattering technique [85].
The two-step mechanism is plausible and helps explain the overestimation of nucleation kinetics predicted by CNT [9,49,50]. Its application is limited to the observation of disordered, dense liquid or clusters in solution, but the definition of “dense liquid” precursor is vague which leads to the lack of insights into the molecular event of crystal nucleation. For example, it fails to explain solvent-dependent polymorphism. The extent of structure order within the dense liquid is unknown or absent, but the recent solution chemistry measurements show clear evidence of a structural link between solution associates and the resultant crystal synthons [13]. Additionally, the liquid–liquid phase separation or oiling-out often displays a size greater than the micron scale, which should not be considered as the intermediate phase of crystal nucleation but provide two composition environments of nucleation [13].
3.2. Pre-Nucleation Cluster Pathways
Studies on (bio)mineralization of, e.g., calcium carbonates [21] and calcium phosphates [86] and calcium sulfate [30] have shown the presence of pre-nucleation clusters (PNCs) in an aqueous solution. Based on the definition by Gebauer and co-workers, PNCs are soluble solute species (no solid-liquid interface) and thermodynamically stable, which exist in both under- and super-saturated solutions [87]. They thus differ from unstable or metastable clusters formed over the course of crystal nucleation assumed by CNT and dense liquids by the two-step mechanism.
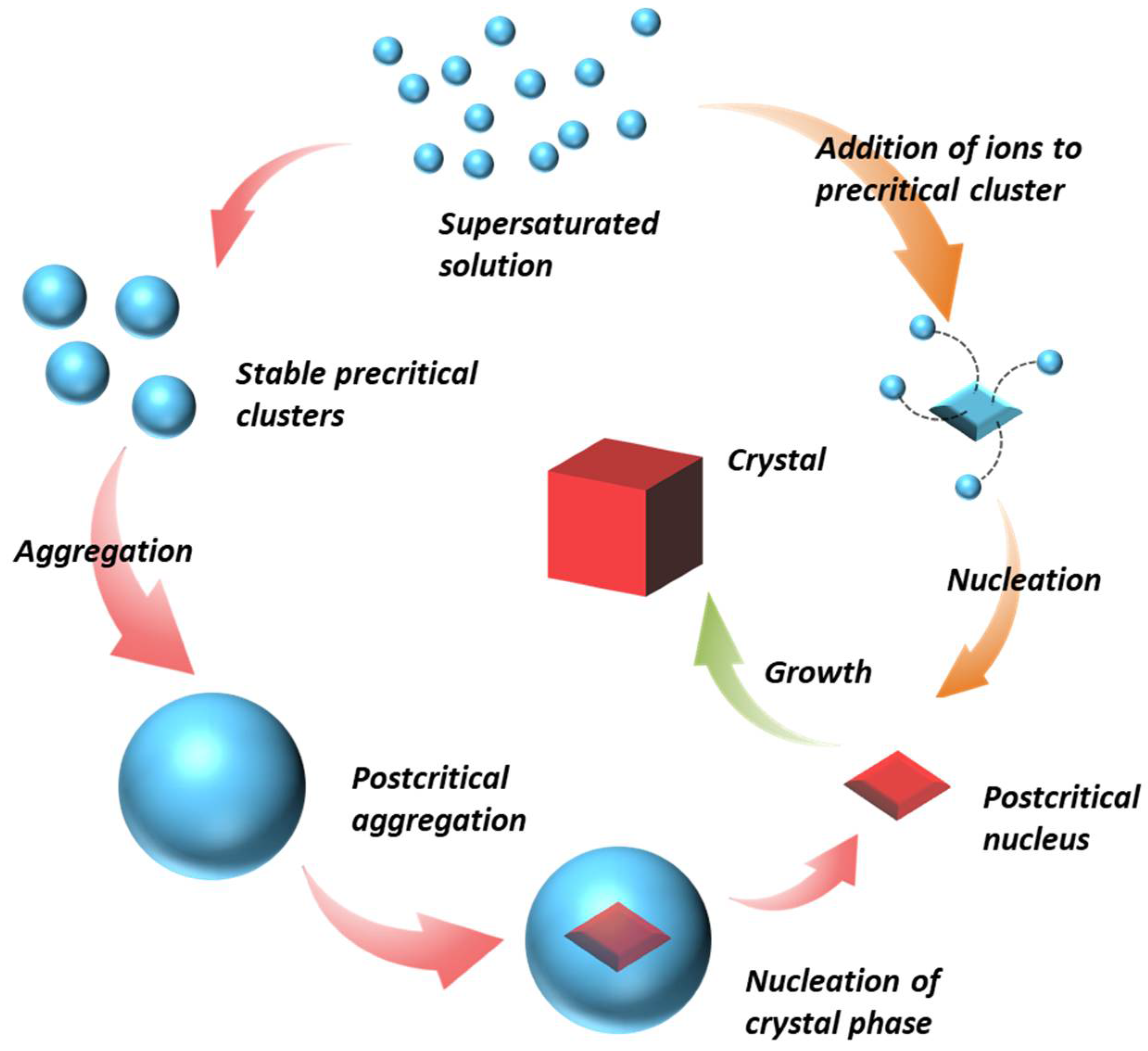
Thermodynamically stable aggregates were found in homogeneous solutions and participated in phase separation in many inorganic systems. Gebauer, D. et al. [21] found the formation of stable ion clusters of calcium carbonate even in unsaturated solutions. They observed the nucleation of amorphous CaCO3 intermediates from a supersaturated solution, which is followed by a phase transition to the crystalline nucleus (Figure 4). More examples of inorganic solutions (e.g., CaSO4, CaPO4) [30,86] and detailed illustrations of PNC pathways can be seen in more recent reports [88,89] and two excellent reviews by Gebauer and Cölfen [14,21].
Although PNC pathways were initially observed in inorganic systems, recent studies present evidence in some organic molecules, mainly amino acids, in aqueous solution. By virtue of electrospray ionization (ESI) and mass spectrometry (MS), Kellermeier et al. [90] found the presence of high-order oligomers of amino acids in a diluted solution. These oligomers were further suggested to be PNCs for the nucleation of amino acid crystals. However, others argued that the observation of oligomers in ESI-MS cannot represent the real situation of oligomerization in an aqueous solution due to different analytical environments. Later on, similar observations of amino acid oligomers were further confirmed by another analytical technique, analytical ultracentrifuge. Molecular-level calculations and simulations [91,92] provide theoretical support for the formation of these oligomers. Interestingly, an AUC analysis of arginine solution [37] showed that the tracked clusters increased as the solution concentration increased toward the saturation limit, and when it reached a certain level, nanoscale populations appeared, then the clusters could no longer be detected in the supersaturated state, which was interpreted as evidence that the amino acid PNC actively participated in the phase separation process.
PNCs provided some evidence that soluble solute oligomers or clusters are present in solution and highlighted the importance of solution chemistry in advancing our understanding of crystal nucleation. In an inorganic system, these aggregates or clusters have been frequently observed experimentally, and their link to amorphous intermediates of nucleation is possible. However, the observations of amorphous intermediates in organic molecular systems like amino acids are relatively rare. Moreover, the thermodynamic stable nature of pre-nucleation clusters in small organic molecular systems is confused. The solute associates in solution should be thermodynamically less stable than monomers due to the interplay of both enthalpy and entropy changes. Indeed, we found solute dimers or even high-order tetramers of benzoic acid can be well explained by the classical molecular self-association model [35].
4. Polymorphism and Molecular Mechanism of Crystal Nucleation
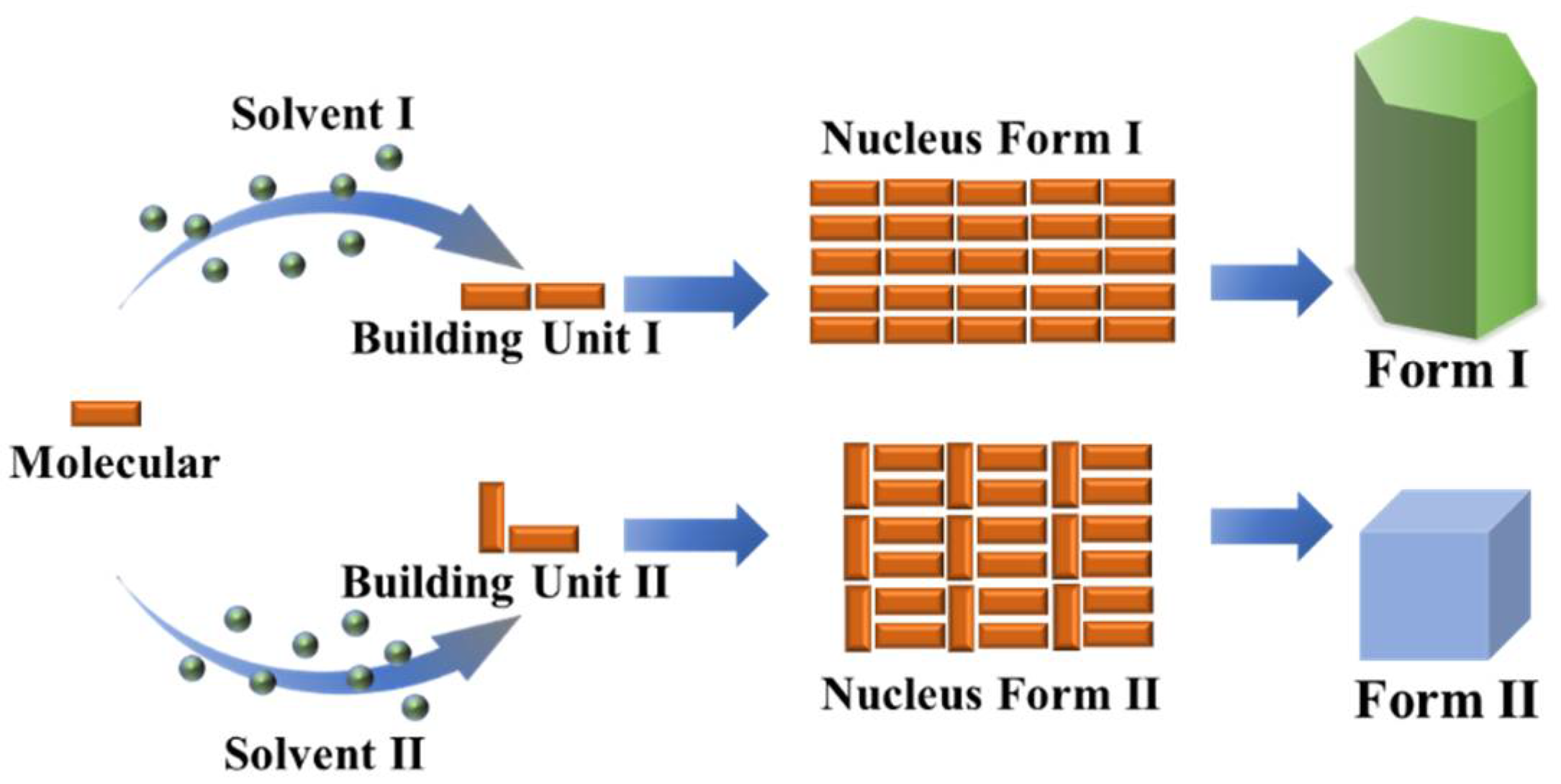
Polymorphism is a ubiquitous phenomenon in many organic crystalline materials. The first definition of polymorphism is given by McCrone [93] as “the possibility of at least two different arrangements of the molecules of a compound in the solid state”. Polymorphs display differences in supramolecular synthons, molecular conformation, intermolecular interactions, molecular arrangements, and different properties. The formation of polymorphs is dictated by the molecular assembly of crystal nucleation but is often explained as a result of the combined effect of crystallization kinetics and thermodynamics [94]. A prominent example was demonstrated recently in protein nucleation by virtue of state-of-art time-resolved cryo-TEM, in which the direct link at the molecular level was established between early-stage nucleation and polymorph selection, and was indicative of a one-step nucleation pathway [31]. The resultant formation of distinct crystal structures in a polymorphic crystallization system could be used as a probe to shed light on the structure perspectives of crystal nucleation [95]. Further, the obtained structure information of crystal nucleation can also be used to test the applicability of classical and non-classical nucleation mechanisms [31].
4.1. Structural Link Correlation between Solution Associates and the Resultant Crystal Synthons
A crystal nucleus is considered as a nanoscale version of macroscopic crystalline materials from a solid-state chemistry perspective and may be also viewed as an amplified assembly of solution chemistry, providing insights into the formation mechanism through a close correlation between the structure synthon and solution building unit. Solution chemistry is an important bridge from molecules to a crystal. Crystallization conditions such as solvent, temperature, supersaturation levels, and physicochemical properties of the solution acting on solute assembly kinetics and solution chemistry have an important influence on crystal nucleation and thus crystallization outcomes [96,97,98,99]. However, the exploration of solution chemistry needs advanced analytical instruments to probe minor populated speciation. The structure of these species is dynamic and often needs to probe using high-precision quantum mechanics (QM) calculations and/or computational simulations. These challenges impeded our understanding of solution chemistry.
With the development and application of advanced solution spectroscopy and computational simulations such as Attenuated Total Reflectance-Fourier Transform Infrared Spectrometer (ATR-FTIR), two-dimensional NOE Nuclear Magnetic Resonance spectroscopy (NOESY), high-accurate QM calculations, and enhanced-sampling molecular dynamic (MD) simulations, solution chemistry is readily available to be explored for probing solute conformation and molecular aggregation (or even clusters) behaviors, as well as their relation with assembly process of crystal nucleation [100,101].
Davey [102] firstly examined the structural connection between solution chemistry and crystal synthons and found that pre-nucleation associates in solution display a certain resemblance to the structural synthon of the resultant crystal phase in a number of organic carboxylic acid systems. The significant similarity in structure correspondence was further revealed by solution IR spectroscopy and computer simulation studies. Such a structural link explains solvent-dependent polymorphism in some cases and also suggests, at least at the dimer level, that the nature of the associate and its intermolecular binding may be an important factor in crystal nucleation. As shown in Figure 5, the self-association in two different solvents leads to the presence of two types of dimers in the solution, eventually resulting in the nucleation of two different polymorphic crystal structures. Hunter et al. [103] examined the assembly of carbamazepine at the dimer level in solution using solution NMR techniques and simulation methods, and dimers of solute molecules assembled by hydrogen-bonding interactions were formed in CDCl3 whereas the stacked dimers by aromatic-aromatic interactions were shown in CD3OH solution. Further examination of the structural correlation of these dimers in solution with structure synthons of carbamazepine polymorph Ⅲ also unveils a remarkable resemblance. By virtue of solvent-dependent self-association of solutes, the formation conditions of new crystalline polymorph may be discovered such as crystallization of isonicotinamide (INA) in chloroform and tetrolic acid [104] in dioxane [105].
On the other hand, examination of solution chemistry and structural synthons of the cocrystal forming system also reveals a similar structure correspondence. A typical case is the binary system of benzophenone (BZP) and diphenylamine (DPA), which can form 1:1 cocrystal, and NMR spectroscopy and chemical shift modeling demonstrated a solvent-dependent effect of solution association, in which hydrogen-bonded dimers produce in toluene and aromatic-aromatic dimers form in methanol. Both associates display excellent structure correspondence with the final cocrystals [106]. Moreover, the extended octameric associates consisting of closed-loop hydrogen-bonded tetramers stacked by π-π interactions in solution were found retained in the resultant crystal structures for a series of aniline-phenol cocrystal systems [107].
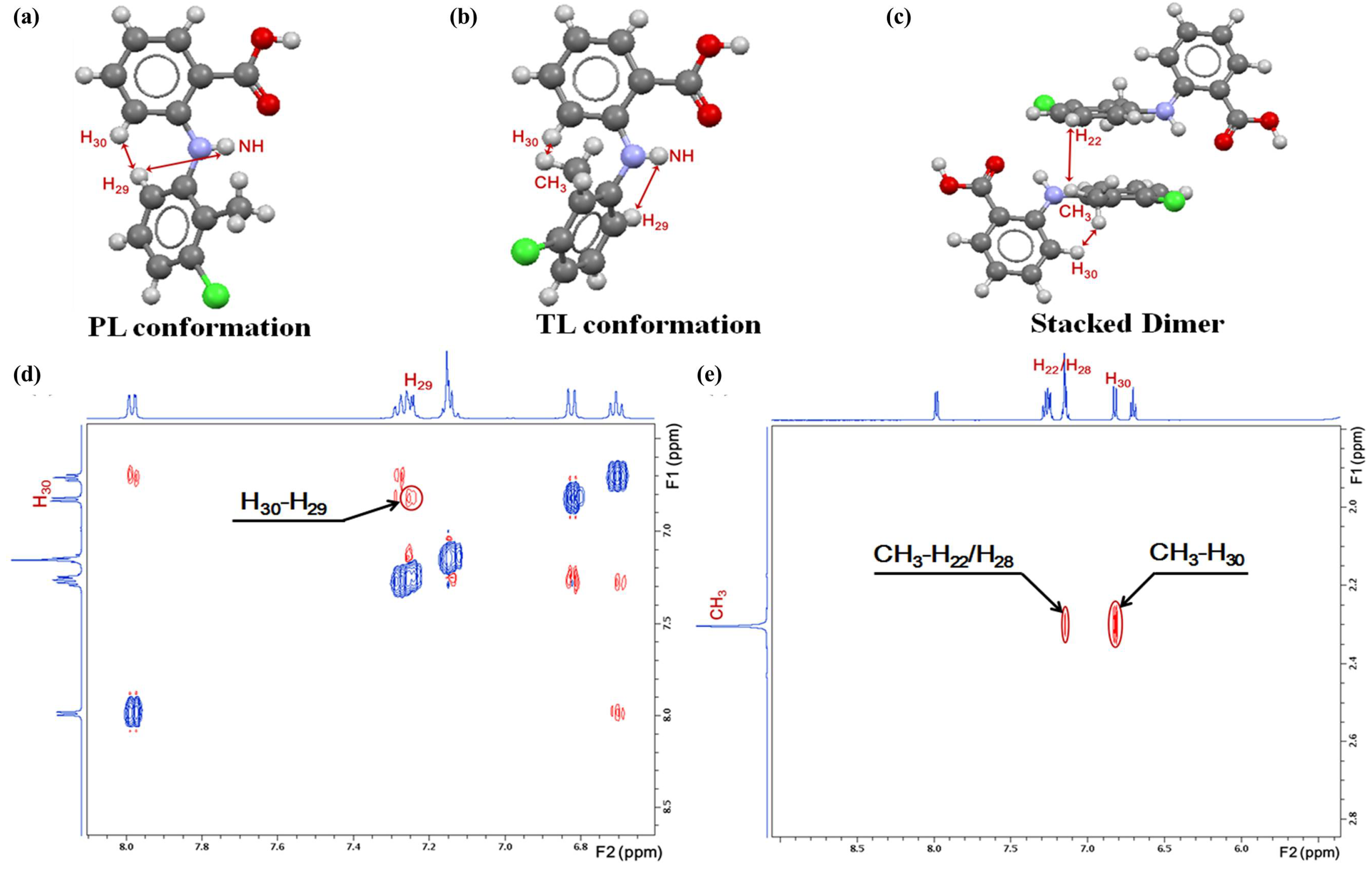
Although the aforementioned structural link can be observed in a number of small organic molecule systems and helps explain polymorphic nucleation, indicating the applicability of CNT at the dimer level, there are many other systems wherein this structure correspondence is absent. We recently studied the solution chemistry of a diarylamine compound, tolfenamic acid (TFA), aiming to examine the roles of molecular conformation and solution association played on pathways of crystal nucleation [108]. TFA is a benchmark conformational polymorph system, showing two distinct conformers, i.e., “twisted-like” (TL) and “planar-like” (PL), in polymorphs I and II, respectively (Figure 6a,b). Both polymorphic structures are composed of hydrogen-bonded, carboxyl homodimers as the supramolecular synthon. Using the 2D NOESY technique, the close spatial approximations of TFA CH3−H30 and H30−H29 protons were observed in ethanol, toluene, and DMF from strongly polar to non-polar solvents, indicating the presence of both conformers in the three selected solution systems that vary in conformational populations. Moreover, the cross-peaks of CH3−H22/H28 protons are also seen in ethanol (Figure 6e) and DMF, demonstrating the self-association of TFA via aromatic stacking interactions (Figure 6c). Indeed, we found the nucleation of DMF solvate crystals of stacked dimer motifs in DMF solvent. The hydrogen-bonded TFA dimer species found in toluene can correlate with their counterparts in the crystal structure. These results corroborate the structure link hypothesis, but the careful examination of TFA associates in ethanolic solution and crystals exhibits an evidently absent correlation. The solvated, stacked TFA-TFA dimers differ from hydrogen-bonded, carboxyl homodimer motifs in crystals suggesting the desolvation and supramolecular reconstruction in the nucleation pathway. The absent structure correspondence was reported in many other systems. For example, the self-associates of different benzoic acid derivatives in polymorphic crystals with different polar solvents do not show a linear correspondence with the final crystal structure [102,109,110,111].
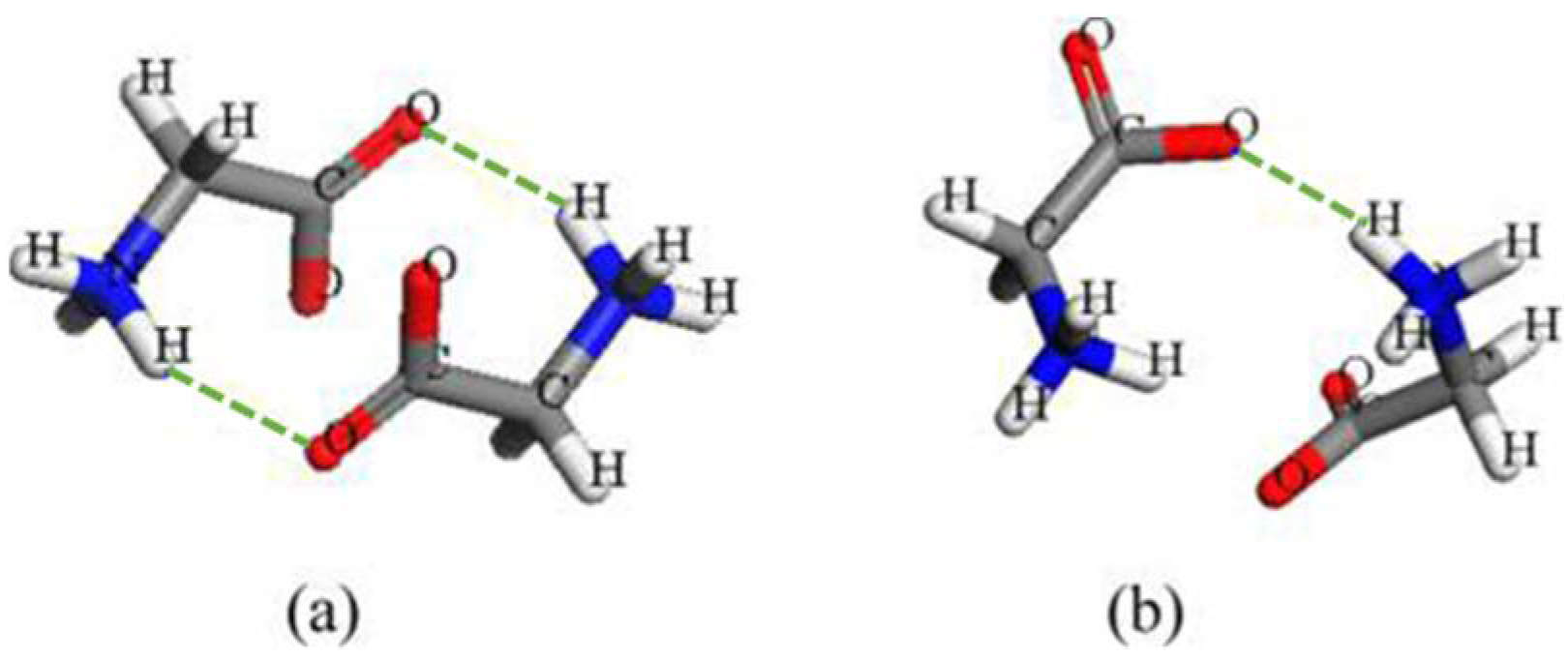
Glycine is one of the representative systems for investigating nucleation which has two different hydrogen-bonded dimers in -their structure (Figure 7) [26,112,113]. In the crystal structure, glycine molecules are packed as cyclic hydrogen-bonded dimers in α polymorph but as a chain hydrogen-bonded dimer in γ form. It was initially postulated that the formation of a cyclic, hydrogen-bonded dimer in water leads to the selective crystallization of the α form [114]. Later on, the amounts of glycine dimers in the water were measured ranging from 5% to 30% (mole fraction) using a series of experimental techniques, including freezing-point depression [115], diffusivity [91], dielectric relaxation [116], and ultracentrifugation [37]. Further computational studies probed structures of various dimers, but the stability or energy ranking of dimer species varied with the use of computation methods [28]. One quantum mechanical study suggested that the cyclic hydrogen-bonded dimer is more stable than the open one [117], whereas molecular dynamic simulations showed that open dimers are more dominant [113]. We investigated glycine polymorphic crystallization using both experiments and simulations, and revealed pH-dependent glycine self-association which correlates with polymorphic formation [114]. By examining the inter-proton contact signal between 12C and 13C methylene protons of an open hydrogen-bonded dimer using 2D NOESY, we are able to unveil the dimer species of glycine, despite its dynamic nature, that are predominantly in open-chain structure. The apparent structural link is thus also absent in glycine polymorphic crystallization.
4.2. Examination of Solution Chemistry with Nucleation Kinetics
A number of experimental techniques for measuring crystal nucleation rates have become available nowadays, such as the double-pulse technique [118,119], microfluidics [120,121], and the high-throughput setup [122,123]. These analytical techniques produce a large number of nucleation kinetics data. The recent advancement in the statistical analysis of measured induction times or nucleation rate by using Poisson distribution explains the rare event of nucleation with the single nucleation model [124]. Other attempts combined high-throughput experimental methods and the statistical analysis of measured induction times to explore the correlation of crystallization conditions [125,126], solvation and dimerization of solute molecules in nucleation assembly [127], molecular attachment frequency and interfacial energy [36,122], and intermolecular interaction energies [128] with nucleation kinetics. In addition, machine learning has recently been applied to the study of nucleation kinetics in some inorganic and colloidal nanoparticle systems to estimate the nucleation rate or the formation rate of precursors [129,130,131].
The first thorough examination of the relationship between solution chemistry and nucleation kinetics was performed by Davey and co-authors [127]. The solution speciation analyses of p-aminobenzoic acids in 2-propanol, acetonitrile, and ethyl acetate revealed the predominant solvated monomers in three solvents wherein strong solvation was suggested in 2-propanol and the least solvation in acetonitrile. The solution speciation of benzoic acid in toluene was used as a reference state where the hydrogen-bonded dimers of benzoic acid were found. They further found that the solvation capability and difficulty of dimerization are inversely correlated with the molecular attachment frequency and nucleation rate, which suggests desolvation and dimerization formation being the rate-determination step of crystal nucleation. However, later studies show that the presence of hydrogen-bonded dimer pre-assembly cannot correlate with fast nucleation kinetics or molecular attachment frequency [36,122]. On the other hand, the solvation capacity does correlate with the difficulty of crystal nucleation [132]. Our recent study [122] suggests that the rate of crystal nucleation is pre-dominant by thermodynamic interface energy rather than kinetic factors, for example, molecular attachment to the critical nucleus.
4.3. Role of Solvation and Aromatic Interactions
Numerous studies have shown the influence of solvent on crystal nucleation, and the solvation effect has been well-recognized [27,127,133]. However, only recently it was examined at the molecular level. It was found the hydrogen-bonded dimer associates are rarely presented in most polar solvents wherein solutes form strong interactions with solvent molecules, which create solvation layers and impedes the self-association of solutes. Indeed, many studies report the solvent dependence of molecular association. We found the self-association of α, ω-alkanedicarboxylic acids absent in, hydrogen-bonding donor (HBD) solvents, but it appears in no-HBD solvent systems [134]. Other studies also observed the formation of only monomers or solvated aromatic stacking dimers in some polar solvents [103,135]. The solvation effect of solutes imposed by solvent molecules has an important influence on solute assembly and affects polymorphic nucleation. It has been demonstrated from both experiments and mathematical modeling that desolvation of solutes in the nucleation pathway results in concurrent nucleation of TFA form I and form II and the appearance of concomitant polymorphism (Figure 8) [136,137]. The solvated monomers may initially form dimers or aggregates through weak intermolecular interactions, e.g., aromatic stacking and/or van der Waals forces, which occurs in either the pre-nucleation or nucleation stage. Then, desolvation of the solutes’ aggregates could be the key step of crystal nucleation, which codes the way of reconstruction and the selectivity of packing arrangements, i.e., polymorphs. The kinetic nature of this desolvation is likely to give rise to the formation of different critical nuclei and the formation of two polymorphs in the same batch. The scenario is similar to the two-step nucleation mechanism regarding the sequential development of density and structural fluctuations but essentially differs in the structure of intermediates. Here the intermediates are soluble aggregated clusters of solvation layers with certain short-range ordering, whereas in the two-step nucleation mechanism they are disordered dense liquids.
Although the above scenario of the desolvation pathway is likely, the particular characteristics of molecular structure may need for step-by-step desolvation due to the binding specificity of interaction sites of solutes with solvent molecules. By using molecular dynamics simulations, Dighe et al. [27] show the sequential and selective desolvation of glutamic acid (GLU) from a supersaturated aqueous solution. Solvation alone could not fully explain complex nucleation behaviors, and another important factor, weak intermolecular interactions, e.g., aromatic interactions, also played an important role in the pathway of crystal nucleation.
The critical importance of aromatic interactions on nucleation pathways may be firstly recognized in explaining unusual concomitant crystallization of TFA polymorphs from ethanolic solution [108]. The strong solvation effect and stacking self-association of TFA were seen in both DMF and ethanol. In DMF, these solvated, stacking associates lead to the direct formation of DFM solvate crystals of TFA with resemble structure motifs of hydrogen-bonded TFA-DMF and aromatic stacking dimer. However, the similar stacking dimers of TFA in ethanol result in the formation of distinct hydrogen-bonded carboxyl homodimers. The observed remarkable difference hints that aromatic interactions of TFA molecules act on the assembly pathway and kinetics of crystal. Later on, the aromatic interactions were found to drive the formation of high-order aggregates in benzoic acid [35] and its derivatives [100].
Not only solution chemistry and thermodynamics, but aromatic interactions are also responsible for kinetics and pathways of crystal nucleation. We recently examined nucleation kinetics of flufenamic acid (FFA) in a series of solvents and found that the breakage of solvent-solute hydrogen bonds and dimerization is not the rate-determination step of crystal nucleation rate. By correlating growth rate with nucleation kinetics, we revealed that weak aromatic interactions affect the nucleation pathway by probably regulating the growth of the nucleating clusters [36]. Cruz-Cabeza et al. [128] through compressive nucleation kinetic measurements and computational simulations demonstrated that the nucleation rates of four benzoic acid derivatives in a series of solvents correlate reasonably well with dimerization energies of aromatic stacks. Rosbottom et al. [138] found that nitromethane (NMe) disrupts π-π interactions by changing the solvent composition to modulate the polycrystalline form of α-p-aminobenzoic acid (pABA), thereby inhibiting the growth in the long-axis direction dominated by π-π interactions. Although these preliminary studies have highlighted the important roles of aromatic interactions in thermodynamics and kinetics of crystal nucleation which affect polymorphic crystallization outcomes, the underlying action mechanism on nucleation pathways remains to be further understood.
4.4. Hierarchical Intermolecular Interactions in Solution Assembly
The formation of a crystalline nucleus may be viewed as a molecular assembly process wherein the building units were constructed according to a certain rule to form a macroscopic crystal. Traditionally, the rule was interpreted as the interplay between thermodynamics and kinetics of crystallization, but the underlay molecular mechanism is lacking and more importantly, this explanation cannot guide the manipulation of crystal synthesis at the molecular level. Understanding such a rule has been a long-standing task for crystal engineers. The great breakthrough was firstly made by Desiraju [139,140,141] who recognized the importance of non-covalent bond interactions, i.e., hydrogen bond, in crystal engineering and proposed the concepts of supra-molecular synthons [142]. The concept receives great attention and now has become a regular tool to guide or predict the synthesis of various crystalline materials. However, other non-covalent bond interactions such as aromatic interactions and van der Waals interactions, beyond hydrogen bond, are still less understood due to their even weak force in essence. Moreover, the synthesis of organic crystalline materials often involves multiple weak interactions [143] and how these interactions aligned to form a crystal are still unclear.
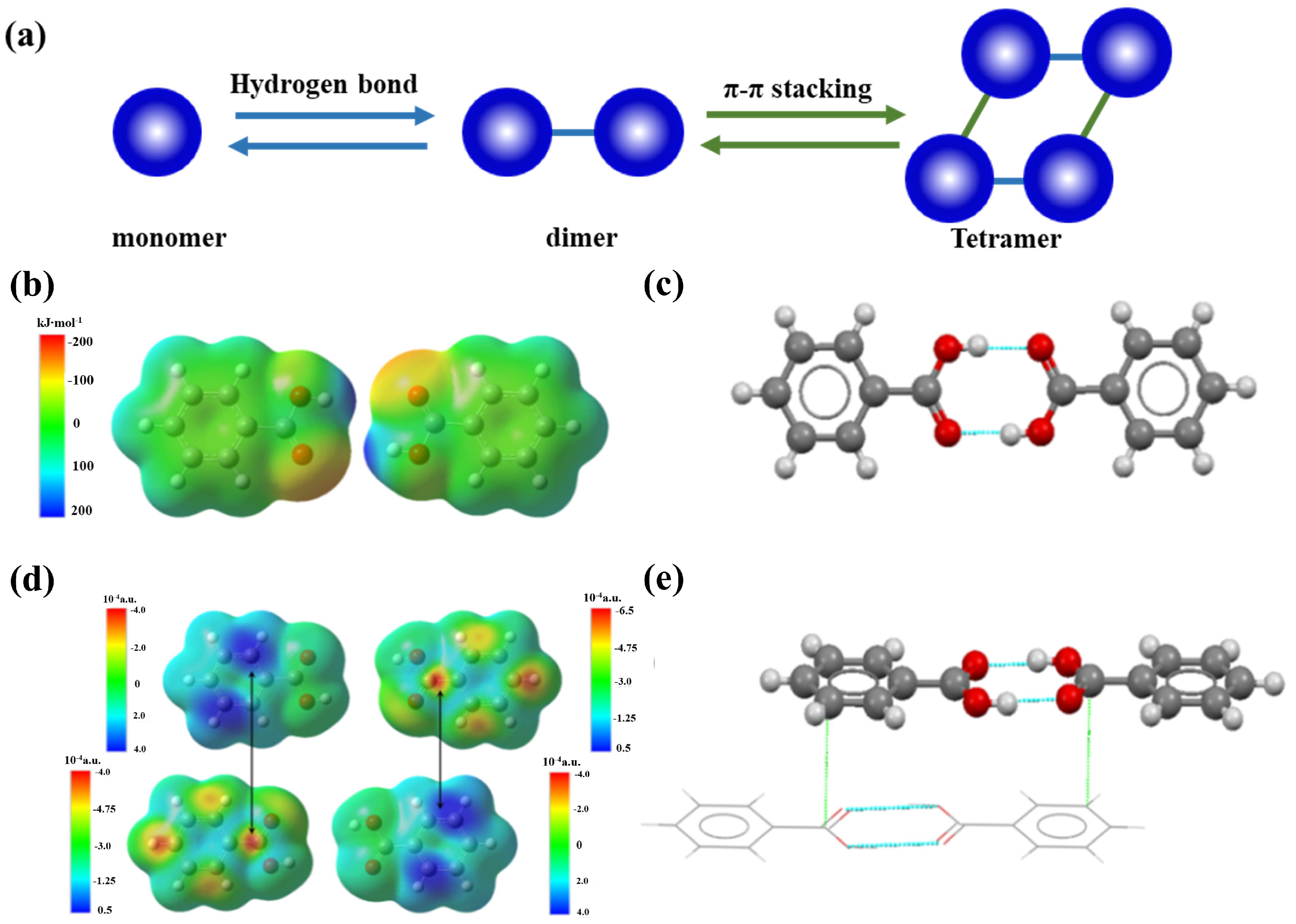
The self-association and aggregation of molecules in solution which precede the nucleation stage has been studied extensively and corroborated at the dimer level [96,144]. However, the large solutes’ aggregates were rarely reported in small organic molecular systems. We recently demonstrated the hierarchical self-assembly of benzoic acid molecules in toluene by forming sequential hydrogen-bond and π⋯π aromatic stacking interactions (Figure 9a) which elucidate the competitive and synergistic nature of multiple intermolecular interactions [35]. At low concentrations, benzoic acid molecules form hydrogen-bonded dimers, whereas high-order tetramers are formed at high concentrations through the stacking of two dimers. The findings hint at hierarchical characteristics of multiple intermolecular interactions in crystal nucleation. The nature of hierarchical interactions was further illustrated by electrostatic potential (ESP) and Fukui functions of monomer and dimer species of BA molecules. The primary hydrogen bond force is formed via mainly electrostatic interactions between carboxylic acid groups, producing cyclic hydrogen-bonding dimers with an energy of −66.5 kJ/mol for two hydrogen bonds (Figure 9b,c). In contrast, the weaker, secondary force, π⋯π interactions, formed through a soft-soft type of interactions that are characterized by matching of Fukui functions have −51.3 kJ/mol for two π⋯π contacts (Figure 9d,e).
Hierarchical intermolecular interactions were also seen in the nucleation of macromolecular systems e.g., proteins. The recently discovered higher-order dimer-based aggregation of actin is observed by the initial formation of homodimers and subsequent tetramers, acting as the primary form of nucleation [145]. In addition, multiple interactions involved in hydrogen bonding, aromatic interactions, and dispersion forces were also investigated to understand the self-assembly of short aromatic peptides, which emphasizes the importance of hierarchical interactions in the rational design and performance control of peptide-based materials [146]. Understanding higher-order self-assembly driven by multiple interactions will help to elucidate the molecular assembly picture of nucleation pathways. The current exploration of hierarchical interactions remains in the nature of solution chemistry and thermodynamics, which does provide important insights into the assembly pathway of crystal nucleation. Further studies on hierarchical interactions may provide structural insights into nucleation intermediates such as clusters, dense liquids, and amorphous phases.
4.5. Aggregation-Based Multiple Nucleation Pathways
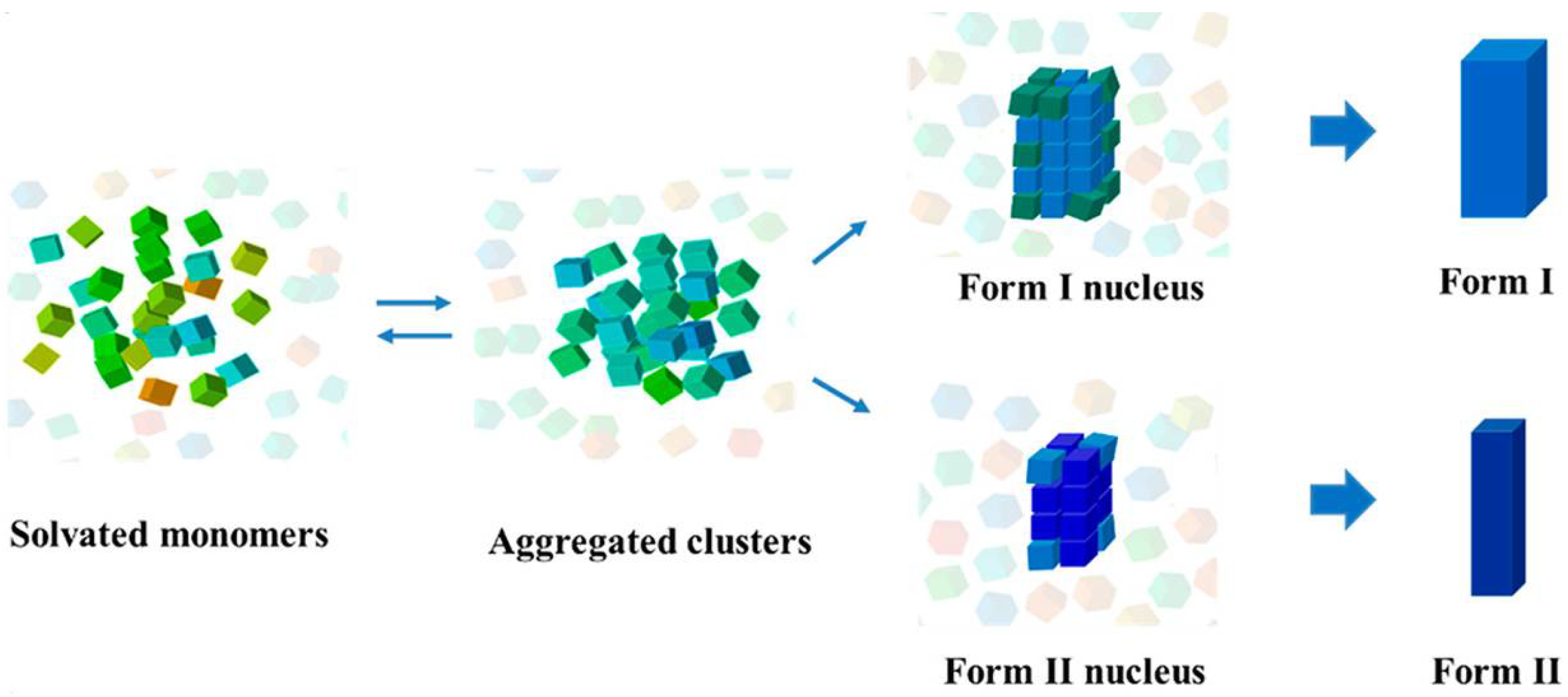
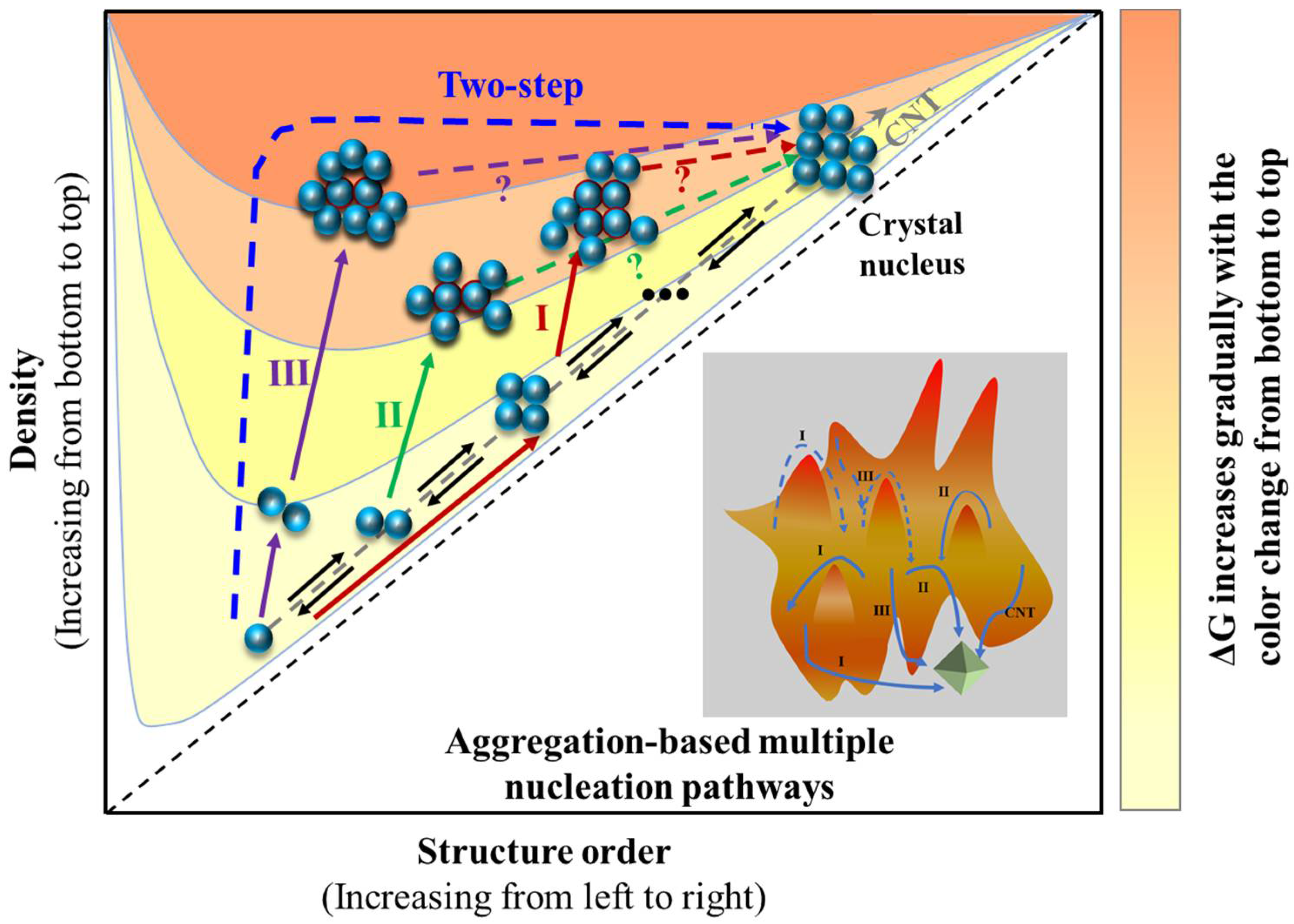
Collectively, the structural link suggests the potential reservation of hydrogen-bonded solute associates in molecular clusters and crystal nuclei [108]. Nevertheless, the hierarchical nature of non-covalent bond interactions including hydrogen bond, aromatic stacking, and van der Waals demonstrates that the high-order structure of these associates should bear certain domains composed of aromatic or van der Waals interactions differing from the crystal nucleus. In other words, the density of high-order associates/aggregates (metastable clusters relative to bulk solution) is developed by molecular association/aggregation likely prior to their structure development in the pre-nucleation stage. The associates or aggregates as illustrated in the aforementioned systems bear the inevitable solvation effect [127,147], and desolvation of solvent molecules [148,149,150] accompanied by superamolecular reconstruction leads to the formation of a nucleus. Recent studies have revealed the detailed kinetics of orientated attachment and the importance of desolvation on particle aggregation during crystal nucleation [80,151]. The large barrier of this step results in the rare event of crystal nucleation, which may also produce simultaneous nucleation of two crystal structures (polymorphs) [152,153,154], i.e., concomitant crystallization of polymorphs when the nucleation barrier for two polymorphs were comparable. Furthermore, the absence of structure correspondence and solvent-dependent molecular association indicate multiple aggregates of solutes regulated by their surrounding environment that is formed either in pre-nucleation or in the course of nucleation stages. The molecular aggregates undergo desolvation and superamolecular reconstruction prior to forming the structure synthon seen in the resultant crystal structure. Again, since the hierarchical nature of non-covalent bond interactions in an organic crystallization system, various molecular aggregates could be formed and thus lead to multiple nucleation pathways, as illustrated in Figure 10. Therefore, we argue that the nucleation pathways of organic molecules are multiple and dependent upon solvent, supersaturation, pH, and solute structure. For example, benzoic acid molecules in toluene initially assemble into hydrogen-bonded dimers and then form high-order tetramers via π⋯π stacking interactions between dimers at high concentrations where the development of crystalline order accompanies the density evolution (pathway I). It is possible that the tetrameters further aggregate to form large clusters via even aromatic weak interactions which become sites of crystal nucleation. Note that here density fluctuations will deviate from the development of crystalline order. The relatively strong hydrogen-bonding and π⋯π stacking interactions are reserved in the resultant crystal structure, but other weak interaction motifs are not. A similar scenario may be envisaged in the case of TFA nucleation from toluene. Instead of the formation of high-order tetramers, the TFA molecules are able to form certain population of hydrogen-bonded dimers, which structurally resemble the resultant crystal structure, but further development of large aggregates or clusters leads to faster development of clusters’ density than crystalline order (pathway II). If solute-solute and solute-solvent interactions are both strong, the solutes may still form self-associates or dimers that are modified by the strong solvation effect. In this scenario, the development of the density of clusters will be much faster than crystalline order (pathway III). The behaviors were seen in several organic molecular systems such as nucleation behaviors of glycine in aqueous solution and TFA in ethanol. If solute-solvent interactions are very strong and solutes themselves are weak, the fully disordered pathway may be seen as suggested in the two-step nucleation mechanism.
Similar multiple nucleation pathways were previously suggested in the colloidal system where the interactions among colloidal spheres are even weaker [155]. The suggested nucleation mechanism may be upset due to the difficult manipulation of weak solute interactions and the design of crystalline materials from molecular building units. However, these multiple nucleation pathways could provide an opportunity to achieve the target crystalline materials via appropriate regulation of molecular aggregates (or clusters) in the pre-nucleation or nucleation stage.
Multiple nucleation pathways were previously suggested by De Yoreo [64] in explaining the nucleation behavior of an inorganic system. Nielsen et al. observed, using in situ liquid TEM, different intermediate phases of CaCO3 in nucleation that gradually evolve from disorder to order phase, indicating multiple nucleation pathways. Kimura et al. used liquid-cell transmission electron microscopy aided by machine learning and observed multiple precursors in the early stages of nucleation [130]. Later on, it was also demonstrated that minor modification of the hydrophobic side chain of peptoid molecules will shift from single-step nucleation to a two-step pathway [156]. Other studies show the change of nucleation pathways by the presence of ions or additives. Amorphous Calcium Carbonate (ACC) is a common form in calcium carbonate crystallization, and the pre-nucleation cluster (Prc) is an intermediate state frequently observed in calcium carbonate crystallization processes. In the system of calcium carbonate solution in the presence of a high concentration of magnesium ions, doping of magnesium ions forms several intermediates of ACC-Mg and Prc-Mg which participate in the nucleation process of calcium carbonate [157]. Citrate is used as a commercial inhibitor for kidney stones consisting of mainly calcium oxalate. Recent studies show citrate altering the nucleation pathway of calcium oxalate hydrates by interacting with pre-nucleation aggregates and amorphous intermediates mainly through water incorporation and local structure ordering [158]. More recently, multiple nucleation pathways are also seen in protein systems [31,80].
Computer simulations could provide additional support [64] for aggregates-based multiple nucleation pathways in both inorganic and organic systems. Nucleation of sodium chloride was traditionally seen by classical nucleation framework, whereas the recent molecular dynamic (MD) simulation studies unveil a much more complex nucleation pathway in which large clusters were observed prior to the appearance of an ordered crystalline phase [159]. Lanaro et al. [160] also used MD simulations to detect the presence of small clusters of small to six ions in saturated solutions of NaCl, which were shown to be potential nuclei for NaCl crystallization.
The aggregation-based multiple nucleation pathways need to be further understood, but they have provided plausible explanations for the polymorphic formation of small organic molecule systems.
5. Challenges and Open Questions
Both classical and non-classical nucleation theories have their own limitations, and there are still lots of questions unanswered. Recent advancements in nucleation rate measurements with statistical analysis have motivated large amounts of nucleation rate measured in a number of organic molecule systems. However, these data appear not very helpful to extract key molecular kinetics information of crystal nucleation. On the other hand, solution spectroscopy techniques have been extensively applied to elucidate structure information of the pre-nucleation or nucleation associates and/or aggregates, providing the structural basis to understand and potentially control polymorphic nucleation at the molecular level. However, more research is needed to understand the detailed molecular mechanism of crystal nucleation in the following areas:
- (1)
- Observing the dynamical structure of a crystal nucleus in small organic molecule systems at the molecular level remains a great challenge, which needs further development of in situ characterization tools (e.g., liquid-cell TEM) for small organic crystalline materials combined with computational simulations to elucidate molecular information of crystal nucleation;
- (2)
- The dispute whether density and structure fluctuations are concomitant remains to be answered between classical and non-classical nucleation mechanisms. Although some computational results support that at certain crystallization conditions crystalline order is preceded by density fluctuations in non-classical nucleation mechanisms, the claim is still lacking experimental evidence in small organic molecule systems;
- (3)
- The nature of stable clusters in the pre-nucleation cluster pathway is unclear, and examinations of the size and structure evolution of these pre-nucleation associates and/or aggregates toward crystal nucleation are critical to providing a deeper fundamental understanding of nucleation. These pre-nucleation solute species are only recently accessible using solution spectroscopy and high-energy x-ray scattering combined with PDF analysis. Moreover, only the size of clusters was considered in CNT, which is unlikely in organic molecule systems;
- (4)
- The predictable and efficient control method for homogenous nucleation is still desired with the tailored properties of crystalline materials. The decisive role of weak interactions for example aromatic interactions and van der Waals forces on crystal nucleation may lead to control nucleation difficult.
6. Summary and Outlook
The future manufacturing of crystalline materials requires the predictable and rational assembly of crystals from molecules to synthesize materials with targeted properties. This needs precise control of the crystallization process, in particular crystal nucleation, based on a deeper fundamental understanding of the nucleation mechanism. Both classical and non-classical nucleation mechanisms lack a detailed elucidation of the assembly information of crystal nucleation. The nature of the solution species that appeared in either pre-nucleation or nucleation stages, for example, pre-nucleation clusters and nucleated intermediates, remain to be understood.
Recent advancements in analytical techniques, measurement methods, and computational simulations have made significant research progress on mechanistic understandings of crystal nucleation. The use of molecular spectroscopy techniques including FTIR, Raman, and NMR advances our understanding of the role of solution chemistry in pre-nucleation and nucleation stages over the course of crystal nucleation. The solvation and aromatic interactions are likely to play a decisive role in the formation of solution associates or aggregates. The hierarchy in intermolecular interactions suggests the structural complexity of these solution pre-assemblies and their dynamic nature in nucleation pathways, which hints at the nucleation of a molecule involved in multiple nucleation pathways dependent upon crystallization conditions, for example, solvent nature. Additionally, numerous nucleation rate data have been reported with more prominent statistics in the literature. However, the interpretation of these kinetics data appears less efficient to obtain detailed information on nucleation assembly. On the other hand, the computational simulations are expected to assist in revealing the details of the organic crystal nucleation process. However, the current computational simulations on nucleation of organic molecules remain challenged by, e.g., small simulation scales accessible and inappropriate application of bulk properties to clusters of nanoscale dimensions. Although significant advancements were made over the past decades, there are still many open questions in which the application of advanced analytical techniques like high-resolution in situ (cryo-) TEM and computational simulations are expected to unveil more detailed assembly information of nucleation of small organic crystalline materials.
Author Contributions
Conceptualization, resources, supervision and funding acquisition, W.T.; investigation, writing—original draft preparation and writing—review and editing, J.Z. and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC was funded by National Natural Science Foundation of China grant number NNSFC 21808159 and Natural Science Foundation of Tianjin grant number 19JCQNJC04800.
Acknowledgments
The authors are grateful to financial support by National Natural Science Foundation of China (NNSFC 21808159) and Natural Science Foundation of Tianjin (19JCQNJC04800).
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Yoreo, J.J.; Gilbert, P.U.P.A.; Sommerdijk, N.A.J.M.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F.; et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, aaa6760. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, R.N. Kinetic approach to the crystallization of magmatic melts. Dokl. Earth Sci. 2007, 412, 144–146. [Google Scholar] [CrossRef]
- Wang, H.; Liang, X.; Xue, D. Geo-inspired crystallization engineering: Multifunctional materials design and fabrication at nanoscale and beyond. Nanotechnology 2020, 31, 414002. [Google Scholar] [CrossRef] [PubMed]
- Uiterwijk, J. Polymerization and crystallization of Snowflake molecules in Domineering. Theor. Comput. Sci. 2016, 644, 143–158. [Google Scholar] [CrossRef]
- Povey, M.J.W. Nucleation in food colloids. J. Chem. Phys. 2016, 145, 211906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef]
- Bolla, G.; Myerson, A.S. SURMOF induced polymorphism and crystal morphological engineering of acetaminophen polymorphs: Advantage of heterogeneous nucleation. CrystEngComm 2018, 20, 2084–2088. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chavez, A.D.; Li, H.F.; Li, H.; Dichtel, W.R.; Bredas, J.L. Nucleation and Growth of Covalent Organic Frameworks from Solution: The Example of COF-5. J. Am. Chem. Soc. 2017, 139, 16310–16318. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrov, V.D.; Pokyntelytsia, O.A.; Sobolev, A.Y. Thermal hysteresis during the melting and crystallization of macroobjects. Tech. Phys. 2017, 62, 741–744. [Google Scholar] [CrossRef]
- Sun, C.; Xue, D. Crystallization: A phase transition process driving by chemical potential decrease. J. Cryst. Growth 2017, 470, 27–32. [Google Scholar] [CrossRef]
- Kashchiev, D. Nucleation: Basic Theory with Applications; Butterworth Heinemann: Oxford, UK, 2000. [Google Scholar]
- Mullin, J.W. 5—Nucleation. In Crystallization, 4th ed.; Mullin, J.W., Ed.; Butterworth-Heinemann: Oxford, UK, 2001; pp. 181–215. [Google Scholar]
- Davey, R.J.; Schroeder, S.L.M.; Ter Horst, J.H. Nucleation of Organic Crystals—A Molecular Perspective. Angew. Chem. Int. Ed. 2013, 52, 2166–2179. [Google Scholar] [CrossRef]
- Gebauer, D.; Kellermeier, M.; Gale, J.D.; Bergström, L.; Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. [Google Scholar] [CrossRef] [Green Version]
- De Yoreo, J.; Whitelam, S. Nucleation in atomic, molecular, and colloidal systems. MRS Bull. 2016, 41, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Myerson, A.S. Particle Engineering: Fundamentals of Particle Formation and Crystal Growth. MRS Bull. 2008, 31, 881–886. [Google Scholar] [CrossRef]
- Kumomi, H. Nucleation of crystallites and their size distribution. Inorg. Mater. 1999, 35, 608–614. [Google Scholar]
- Li, L.; Reich, S.; Robertson, J. Modelling the Nucleation and Chirality Selection of Carbon Nanotubes. J. Nanosci. Nanotechnol. 2006, 6, 1290–1297. [Google Scholar] [CrossRef]
- Kashchiev, D.; van Rosmalen, G.M. Review: Nucleation in solutions revisited. Cryst. Res. Technol. 2003, 38, 555–574. [Google Scholar] [CrossRef]
- Vekilov, P.G. Nucleation. Cryst. Growth Des. 2010, 10, 5007–5019. [Google Scholar] [CrossRef]
- Gebauer, D.; Cölfen, H. Prenucleation clusters and non-classical nucleation. Nano Today 2011, 6, 564–584. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, D.; Gale, J.D.; Cölfen, H. Crystal Nucleation and Growth of Inorganic Ionic Materials from Aqueous Solution: Selected Recent Developments, and Implications. Small 2022, 2107735. [Google Scholar] [CrossRef]
- Anwar, J.; Zahn, D. Polymorphic phase transitions: Macroscopic theory and molecular simulation. Adv. Drug Deliv. Rev. 2017, 117, 47–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosso, G.C.; Chen, J.; Cox, S.; Fitzner, M.; Pedevilla, P.; Zen, A.; Michaelides, A. Crystal Nucleation in Liquids: Open Questions and Future Challenges in Molecular Dynamics Simulations. Chem. Rev. 2016, 116, 7078–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleutel, M.; Van Driessche, A.E.S. Nucleation of protein crystals—A nanoscopic perspective. Nanoscale 2018, 10, 12256–12267. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Trout, B.L. A Computational Study of the Mechanism of the Selective Crystallization of α- and β-GIycine from Water and Methanol-Water Mixture. J. Phys. Chem. B 2010, 114, 13764–13772. [Google Scholar] [CrossRef]
- Dighe, A.V.; Coliaie, P.; Podupu, P.K.R.; Singh, M.R. Selective desolvation in two-step nucleation mechanism steers crystal structure formation. Nanoscale 2022, 14, 1723–1732. [Google Scholar] [CrossRef]
- Parks, C.; Koswara, A.; DeVilbiss, F.; Tung, H.-H.; Nere, N.K.; Bordawekar, S.; Nagy, Z.K.; Ramkrishna, D. Solubility curves and nucleation rates from molecular dynamics for polymorph prediction—Moving beyond lattice energy minimization. Phys. Chem. Chem. Phys. 2017, 19, 5285–5295. [Google Scholar] [CrossRef]
- Avaro, J.T.; Wolf, S.L.P.; Hauser, K.; Gebauer, D. Stable Prenucleation Calcium Carbonate Clusters Define Liquid–Liquid Phase Separation. Angew. Chem. Int. Ed. 2020, 59, 6155–6159. [Google Scholar] [CrossRef]
- Stawski, T.M.; van Driessche, A.E.; Ossorio, M.; Rodriguez-Blanco, J.D.; Besselink, R.; Benning, L.G. Formation of calcium sulfate through the aggregation of sub-3 nanometre primary species. Nat. Commun. 2016, 7, 11177. [Google Scholar] [CrossRef] [Green Version]
- Van Driessche, A.E.S.; Van Gerven, N.; Bomans, P.H.H.; Joosten, R.R.M.; Friedrich, H.; Gil, D.; Sommerdijk, N.A.J.M.; Sleutel, M. Molecular nucleation mechanisms and control strategies for crystal polymorph selection. Nature 2018, 556, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Pan, A.C.; Rappl, T.J.; Chandler, D.; Balsara, N.P. Neutron Scattering and Monte Carlo Determination of the Variation of the Critical Nucleus Size with Quench Depth. J. Phys. Chem. B 2006, 110, 3692–3696. [Google Scholar] [CrossRef]
- He, Y.; Li, B.; Wang, C.; Mao, S.X. Direct observation of dual-step twinning nucleation in hexagonal close-packed crystals. Nat. Commun. 2020, 11, 2483. [Google Scholar] [CrossRef]
- Urquidi, O.; Brazard, J.; LeMessurier, N.; Simine, L.; Adachi, T.B.M. In situ optical spectroscopy of crystallization: One crystal nucleation at a time. Proc. Natl. Acad. Sci. USA 2022, 119, e2122990119. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, M.; Mo, H.; Gong, J.; Wang, J.; Li, T. Higher-order self-assembly of benzoic acid in solution. Cryst. Growth Des. 2017, 17, 5049–5053. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, S.; Zhang, X.; Tang, W.; Gong, J. Unveiling the Critical Roles of Aromatic Interactions in the Crystal Nucleation Pathway of Flufenamic Acid. Cryst. Growth Des. 2019, 19, 7175–7184. [Google Scholar] [CrossRef]
- Kellermeier, M.; Rosenberg, R.; Moise, A.; Anders, U.; Przybylski, M.; Cölfen, H. Amino acids form prenucleation clusters: ESI-MS as a fast detection method in comparison to analytical ultracentrifugation. Faraday Discuss. 2012, 159, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Erdemir, D.; Evans, J.M.B.; Ilavsky, J.; Amenitsch, H.; Segre, C.U.; Myerson, A.S. SAXS Study of the Nucleation of Glycine Crystals from a Supersaturated Solution. Cryst. Growth Des. 2005, 5, 523–527. [Google Scholar] [CrossRef]
- Baumgartner, J.; Dey, A.; Bomans, P.H.H.; Le Coadou, C.; Fratzl, P.; Sommerdijk, N.A.J.M.; Faivre, D. Nucleation and growth of magnetite from solution. Nat. Mater. 2013, 12, 310–314. [Google Scholar] [CrossRef]
- Tsarfati, Y.; Rosenne, S.; Weissman, H.; Shimon, L.J.W.; Gur, D.; Palmer, B.A.; Rybtchinski, B. Crystallization of Organic Molecules: Nonclassical Mechanism Revealed by Direct Imaging. ACS Cent. Sci. 2018, 4, 1031–1036. [Google Scholar] [CrossRef] [Green Version]
- Houben, L.; Weissman, H.; Wolf, S.G.; Rybtchinski, B. A mechanism of ferritin crystallization revealed by cryo-STEM tomography. Nature 2020, 579, 540–543. [Google Scholar] [CrossRef]
- Van Driessche, A.E.S.; Ling, W.L.; Schoehn, G.; Sleutel, M. Nucleation of glucose isomerase protein crystals in a nonclassical disguise: The role of crystalline precursors. Proc. Natl. Acad. Sci. USA 2022, 119, e2108674119. [Google Scholar] [CrossRef]
- Chung, S.; Shin, S.-H.; Bertozzi, C.R.; De Yoreo, J.J. Self-catalyzed growth of S layers via an amorphous-to-crystalline transition limited by folding kinetics. Proc. Natl. Acad. Sci. USA 2010, 107, 16536–16541. [Google Scholar] [CrossRef] [Green Version]
- Sleutel, M.; Lutsko, J.; Van Driessche, A.E.; Durán-Olivencia, M.A.; Maes, D. Observing classical nucleation theory at work by monitoring phase transitions with molecular precision. Nat. Commun. 2014, 5, 5598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelton, K.F.; Frenkel, D. Preface: Special Topic on Nucleation: New Concepts and Discoveries. J. Chem. Phys. 2016, 145, 211501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, H.-H. Crystallization of Organic Compounds: An Industrial Perspective; John Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- Bai, G.; Gao, D.; Liu, Z.; Zhou, X.; Wang, J. Probing the critical nucleus size for ice formation with graphene oxide nanosheets. Nature 2019, 576, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Kashchiev, D. Analysis of experimental data for the nucleation rate of water droplets. J. Chem. Phys. 2006, 125, 044505. [Google Scholar] [CrossRef]
- Sharaf, M.A.; Dobbins, R.A. A comparison of measured nucleation rates with the predictions of several theories of homogeneous nucleation. J. Chem. Phys. 1982, 77, 1517–1526. [Google Scholar] [CrossRef]
- Strey, R.; Wagner, P.E.; Schmeling, T. Homogeneous nucleation rates for n-alcohol vapors measured in a two-piston expansion chamber. J Chem Phys. 1986, 17. [Google Scholar] [CrossRef]
- Schmelzer, J.W.; Abyzov, A.S. Crystallization of glass-forming liquids: Thermodynamic driving force. J. Non-Cryst. Solids 2016, 449, 41–49. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A. Crystallization of glass-forming liquids: Specific surface energy. J. Chem. Phys. 2016, 145, 064512. [Google Scholar] [CrossRef]
- Espinosa, J.R.; Vega, C.; Valeriani, C.; Sanz, E. Seeding approach to crystal nucleation. J. Chem. Phys. 2016, 144, 034501. [Google Scholar] [CrossRef] [Green Version]
- Tipeev, A.O.; Zanotto, E.D.; Rino, J.P. Diffusivity, Interfacial Free Energy, and Crystal Nucleation in a Supercooled Lennard-Jones Liquid. J. Phys. Chem. C 2018, 122, 28884–28894. [Google Scholar] [CrossRef]
- Gidalevitz, D.; Feidenhans’L, R.; Matlis, S.; Smilgies, D.-M.; Christensen, M.J.; Leiserowitz, L. Monitoring In Situ Growth and Dissolution of Molecular Crystals: Towards Determination of the Growth Units. Angew. Chem. Int. Ed. 1997, 36, 955–959. [Google Scholar] [CrossRef]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable Prenucleation Calcium Carbonate Clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [Green Version]
- Myerson, A.S.; Trout, B.L. Nucleation from Solution. Science 2013, 341, 855–856. [Google Scholar] [CrossRef]
- Fokin, V.M.; Abyzov, A.S.; Yuritsyn, N.S.; Schmelzer, J.W.; Zanotto, E.D. Effect of structural relaxation on crystal nucleation in glasses. Acta Mater. 2021, 203, 116472. [Google Scholar] [CrossRef]
- Rodrigues, L.R.; Abyzov, A.S.; Fokin, V.M.; Schmelzer, J.W.; Zanotto, E.D. Relaxation effect on crystal nucleation in a glass unveiled by experimental, numerical, and analytical approaches. Acta Mater. 2022, 223, 117458. [Google Scholar] [CrossRef]
- Xia, X.; Van Hoesen, D.C.; McKenzie, M.E.; Youngman, R.E.; Kelton, K.F. Low-temperature nucleation anomaly in silicate glasses shown to be artifact in a 5BaO·8SiO2 glass. Nat. Commun. 2021, 12, 2026. [Google Scholar] [CrossRef]
- Erdemir, D.; Lee, A.Y.; Myerson, A.S. Nucleation of Crystals from Solution: Classical and Two-Step Models. Acc. Chem. Res. 2009, 42, 621–629. [Google Scholar] [CrossRef]
- Rao, A.; Cölfen, H. New Perspectives on Mineral Nucleation and Growth; Springer International Publishing: Berlin, Germany, 2017. [Google Scholar]
- Nielsen, M.H.; Aloni, S.; De Yoreo, J.J. In situ TEM imaging of CaCO3 nucleation reveals coexistence of direct and indirect pathways. Science 2014, 345, 1158–1162. [Google Scholar] [CrossRef]
- De Yoreo, J. Crystal nucleation: More than one pathway. Nat. Mater. 2013, 12, 284–285. [Google Scholar] [CrossRef]
- Russo, J.; Tanaka, H. The microscopic pathway to crystallization in supercooled liquids. Sci. Rep. 2012, 2, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebauer, D.; Gunawidjaja, P.N.; Ko, J.Y.P.; Bacsik, Z.; Aziz, B.; Liu, L.; Hu, Y.; Bergström, L.; Tai, C.W.; Sham, T.K.; et al. Proto-Calcite and Proto-Vaterite in Amorphous Calcium Carbonates. Angew. Chem. Int. Ed. 2010, 49, 8889–8891. [Google Scholar] [CrossRef]
- Vekilov, P.G. Nonclassical Nucleation; American Chemical Society: Washington, DC, USA, 2020. [Google Scholar]
- Ten Wolde, P.R.; Frenkel, D. Enhancement of Protein Crystal Nucleation by Critical Censity Fluctuations. Science 1997, 277, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Talanquer, V.; Oxtoby, D.W. Crystal nucleation in the presence of a metastable critical point. J. Chem. Phys. 1998, 109, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.H.; Liu, X.Y. How Does a Transient Amorphous Precursor Template Crystallization. J. Am. Chem. Soc. 2007, 129, 13520–13526. [Google Scholar] [CrossRef] [PubMed]
- Galkin, O.; Pan, W.; Filobelo, L.; Hirsch, R.E.; Nagel, R.L.; Vekilov, P.G. Two-Step Mechanism of Homogeneous Nucleation of Sickle Cell Hemoglobin Polymers. Biophys. J. 2007, 93, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vekilov, P.G. Dense Liquid Precursor for the Nucleation of Ordered Solid Phases from Solution. Cryst. Growth Des. 2004, 4, 671–685. [Google Scholar] [CrossRef]
- Wallace, A.F.; Hedges, L.O.; Fernandez-Martinez, A.; Raiteri, P.; Gale, J.D.; Waychunas, G.A.; Whitelam, S.; Banfield, J.F.; De Yoreo, J.J. Microscopic Evidence for Liquid-Liquid Separation in Supersaturated CaCO3 Solutions. Science 2013, 341, 885–889. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Roosen-Runge, F.; Zhang, F.; Roth, R.; Skoda, M.W.A.; Jacobs, R.M.J.; Sztucki, M.; Schreiber, F. Effective interactions in protein–salt solutions approaching liquid–liquid phase separation. J. Mol. Liq. 2014, 200, 20–27. [Google Scholar] [CrossRef]
- Zhang, F.; Roosen-Runge, F.; Sauter, A.; Roth, R.; Skoda, M.W.A.; Jacobs, R.M.J.; Sztucki, M.; Schreiber, F. The role of cluster formation and metastable liquid—liquid phase separation in protein crystallization. Faraday Discuss. 2012, 159, 313–325. [Google Scholar] [CrossRef]
- Braun, M.K.; Wolf, M.; Matsarskaia, O.; Da Vela, S.; Roosen-Runge, F.; Sztucki, M.; Roth, R.; Zhang, F.; Schreiber, F. Strong Isotope Effects on Effective Interactions and Phase Behavior in Protein Solutions in the Presence of Multivalent Ions. J. Phys. Chem. B 2017, 121, 1731–1739. [Google Scholar] [CrossRef]
- Sauter, A.; Roosen-Runge, F.; Zhang, F.; Lotze, G.; Feoktystov, A.; Jacobs, R.M.J.; Schreiber, F. On the question of two-step nucleation in protein crystallization. Faraday Discuss. 2015, 179, 41–58. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Kolomeisky, A.B.; Vekilov, P.G. Nucleation of ordered solid phases of proteins via a disordered high-density state: Phenomenological approach. J. Chem. Phys. 2005, 122, 174905. [Google Scholar] [CrossRef]
- Galkin, O.; Vekilov, P.G. Control of protein crystal nucleation around the metastable liquid–liquid phase boundary. Proc. Natl. Acad. Sci. USA 2000, 97, 6277–6281. [Google Scholar] [CrossRef] [Green Version]
- Van Driessche, A.E.S.; Van Gerven, N.; Joosten, R.R.M.; Ling, W.L.; Bacia, M.; Sommerdijk, N.; Sleutel, M. Nucleation of protein mesocrystals via oriented attachment. Nat. Commun. 2021, 12, 3902. [Google Scholar] [CrossRef]
- Lutsko, J.F.; Nicolis, G. Theoretical Evidence for a Dense Fluid Precursor to Crystallization. Phys. Rev. Lett. 2006, 96, 046102. [Google Scholar] [CrossRef] [Green Version]
- Lutsko, J.F. How crystals form: A theory of nucleation pathways. Sci. Adv. 2019, 5, eaav7399. [Google Scholar] [CrossRef] [Green Version]
- Gavezzotti, A. Molecular Aggregation of Acetic Acid in a Carbon Tetrachloride Solution: A Molecular Dynamics Study with a View to Crystal Nucleation. Chem.-Eur. J. 1999, 5, 567. [Google Scholar] [CrossRef]
- Garetz, B.A.; Matic, J.; Myerson, A.S. Polarization Switching of Crystal Structure in the Nonphotochemical Light-Induced Nucleation of Supersaturated Aqueous Glycine Solutions. Phys. Rev. Lett. 2002, 89, 175501. [Google Scholar] [CrossRef]
- Kovalchuk, M.V.; Blagov, A.E.; Dyakova, Y.A.; Gruzinov, A.Y.; Marchenkova, M.; Peters, G.S.; Pisarevsky, Y.V.; Timofeev, V.; Volkov, V.V. Investigation of the Initial Crystallization Stage in Lysozyme Solutions by Small-Angle X-ray Scattering. Cryst. Growth Des. 2016, 16, 1792–1797. [Google Scholar] [CrossRef]
- Dey, A.; Bomans, P.; Muller, F.; Will, J.; Frederik, P.; De With, G.; Sommerdijk, N. The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat. Mater. 2010, 9, 1010–1014. [Google Scholar] [CrossRef]
- Kellermeier, M.; Gebauer, D.; Melero-García, E.; Drechsler, M.; Talmon, Y.; Kienle, L.; Cölfen, H.; García-Ruiz, J.M.; Kunz, W. Colloidal Stabilization of Calcium Carbonate Prenucleation Clusters with Silica. Adv. Funct. Mater. 2012, 22, 4301–4311. [Google Scholar] [CrossRef] [Green Version]
- Stawski, T.M.; Besselink, R.; Chatzipanagis, K.; Hövelmann, J.; Benning, L.G.; Van Driessche, A.E.S. Nucleation Pathway of Calcium Sulfate Hemihydrate (Bassanite) from Solution: Implications for Calcium Sulfates on Mars. J. Phys. Chem. C 2020, 124, 8411–8422. [Google Scholar] [CrossRef] [Green Version]
- Stawski, T.M.; Van Driessche, A.E.S.; Besselink, R.; Byrne, E.H.; Raiteri, P.; Gale, J.D.; Benning, L.G. The Structure of CaSO4 Nanorods: The Precursor of Gypsum. J. Phys. Chem. C 2019, 123, 23151–23158. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.K.; Fenn, J.B. Formation of charged clusters during electrospray ionization of organic solute species. Biol. Mass Spectrom. 1991, 26, 542–549. [Google Scholar] [CrossRef]
- Hamad, S.; Hughes, C.E.; Catlow, C.R.A.; Harris, K.D.M. Clustering of Glycine Molecules in Aqueous Solution Studied by Molecular Dynamics Simulation. J. Phys. Chem. B 2008, 112, 7280–7288. [Google Scholar] [CrossRef]
- Raiteri, P.; Demichelis, R.; Gale, J.D.; Kellermeier, M.; Gebauer, D.; Quigley, D.; Wright, L.B.; Walsh, T.R. Exploring the influence of organic species on pre- and post-nucleation calcium carbonate. Faraday Discuss. 2012, 159, 61–85. [Google Scholar] [CrossRef] [Green Version]
- Haleblian, J.; McCrone, W. Pharmaceutical Applications of Polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Otto, D.P.; De Villiers, M.M. Solid State Concerns During Drug Discovery and Development: Thermodynamic and Kinetic Aspects of Crystal Polymorphism and the Special Cases of Concomitant Polymorphs, Co-Crystals and Glasses. Curr. Drug Discov. Technol. 2017, 14, 72–105. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.K.; Aggarwal, H.; Nangia, A. Crystal Structure of Methyl Paraben Polymorph II. Cryst. Growth Des. 2011, 11, 967–971. [Google Scholar] [CrossRef]
- Du, W.; Bai, L.; Gong, Y.; Zhang, L.; Xiang, J.; Wang, S.; Tang, N.; Zhu, L.; Yin, Q. Molecular Self-assembly in Solution and the Nucleation Pathway: The Case of p-Nitrobenzoic Acid. Ind. Eng. Chem. Res. 2019, 58, 23284–23293. [Google Scholar] [CrossRef]
- Dimitrov, I.L. Kinetic factors may reshape the dependence of crystal nucleation rate on temperature in protein bulk solution. J. Biol. Phys. 2020, 46, 343–350. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, Z.; Dang, L.; Wei, H. Effect of supersaturation on l-glutamic acid polymorphs under droplet-based microchannels. J. Cryst. Growth 2016, 446, 68–73. [Google Scholar] [CrossRef]
- Jia, L.; Yin, Q.; Zhou, L.; Zhang, X.; Wang, C.; Du, W.; Zhou, L. Insights into the mechanism of concomitant nucleation of form II and ethanol solvate of spironolactone in cooling crystallization. RSC Adv. 2018, 8, 9697–9706. [Google Scholar] [CrossRef] [Green Version]
- Gaines, E.; Maisuria, K.; Di Tommaso, D. The role of solvent in the self-assembly of m-aminobenzoic acid: A density functional theory and molecular dynamics study. CrystEngComm 2016, 18, 2937–2948. [Google Scholar] [CrossRef]
- Zhang, Z.; Walsh, M.R.; Guo, G.-J. Microcanonical molecular simulations of methane hydrate nucleation and growth: Evidence that direct nucleation to sI hydrate is among the multiple nucleation pathways. Phys. Chem. Chem. Phys. 2015, 17, 8870–8876. [Google Scholar] [CrossRef]
- Berzins, A.; Semjonova, A.; Actiņš, A.; Salvalaglio, M. Speciation of Substituted Benzoic Acids in Solution: Evaluation of Spectroscopic and Computational Methods for the Identification of Associates and Their Role in Crystallization. Cryst. Growth Des. 2021, 21, 4823–4836. [Google Scholar] [CrossRef]
- Hunter, C.A.; McCabe, J.F.; Spitaleri, A. Solvent effects of the structures of prenucleation aggregates of carbamazepine. CrystEngComm 2012, 14, 7115–7117. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; McGarrity, E.S.; Meekes, H.; ter Horst, J.H. Isonicotinamide self-association: The link between solvent and polymorph nucleation. Chem. Commun. 2012, 48, 4983–4985. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Trout, B.L. Computational Study of Solvent Effects on the Molecular Self-Assembly of Tetrolic Acid in Solution and Implications for the Polymorph Formed from Crystallization. J. Phys. Chem. B 2008, 112, 7794–7802. [Google Scholar] [CrossRef]
- Chadwick, K.; Davey, R.J.; Dent, G.; Pritchard, R.G.; Hunter, C.A.; Musumeci, D. Cocrystallization: A Solution Chemistry Perspective and the Case of Benzophenone and Diphenylamine. Cryst. Growth Des. 2009, 9, 1990–1999. [Google Scholar] [CrossRef]
- Mukherjee, A.; Dixit, K.; Sarma, S.P.; Desiraju, G.R. Aniline–phenol recognition: From solution through supramolecular synthons to cocrystals. IUCrJ 2014, 1, 228–239. [Google Scholar] [CrossRef]
- Tang, W.; Mo, H.; Zhang, M.; Parkin, S.; Gong, J.; Wang, J.; Li, T. Persistent Self-Association of Solute Molecules in Solution. J. Phys. Chem. B 2017, 121, 10118–10124. [Google Scholar] [CrossRef]
- Davey, R.J.; Dent, G.; Mughal, R.K.; Parveen, S. Concerning the Relationship between Structural and Growth Synthons in Crystal Nucleation: Solution and Crystal Chemistry of Carboxylic Acids as Revealed through IR Spectroscopy. Cryst. Growth Des. 2006, 6, 1788–1796. [Google Scholar] [CrossRef]
- Burton, R.C.; Ferrari, E.S.; Davey, R.J.; Finney, J.L.; Bowron, D. The Relationship between Solution Structure and Crystal Nucleation: A Neutron Scattering Study of Supersaturated Methanolic Solutions of Benzoic Acid. J. Phys. Chem. B 2010, 114, 8807–8816. [Google Scholar] [CrossRef]
- Chiarella, R.A.; Gillon, A.L.; Burton, R.C.; Davey, R.J.; Sadiq, G.; Auffret, A.; Cioffi, M.; Hunter, C.A. The nucleation of inosine: The impact of solution chemistry on the appearance of polymorphic and hydrated crystal forms. Faraday Discuss. 2007, 136, 179–193. [Google Scholar] [CrossRef]
- Shur, V.Y.; Bykov, D.A.; Romanyuk, K.; Rumyantsev, E.L.; Kadushnikov, R.M.; Mizgulin, V.V.; Hosseini, E.S.; Kholkin, A. Formation of self-assembled pattern of glycine microcrystals: Experiment and computer simulation. Ferroelectrics 2016, 496, 20–27. [Google Scholar] [CrossRef]
- Yani, Y.; Chow, P.S.; Tan, R.B.H. Glycine Open Dimers in Solution: New Insights into α-Glycine Nucleation and Growth. Cryst. Growth Des. 2012, 12, 4771–4778. [Google Scholar] [CrossRef]
- Tang, W.; Mo, H.; Zhang, M.; Gong, J.; Wang, J.; Li, T. Glycine’s pH-dependent polymorphism: A perspective from self-association in solution. Cryst. Growth Des. 2017, 17, 5028–5033. [Google Scholar] [CrossRef]
- Huang, J.; Stringfellow, T.C.; Yu, L. Glycine Exists Mainly as Monomers, Not Dimers, in Supersaturated Aqueous Solutions: Implications for Understanding Its Crystallization and Polymorphism. J. Am. Chem. Soc. 2008, 130, 13973–13980. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Buchner, R.; Fernandez, Š.; Chiba, A.; Kunz, W. Dielectric relaxation spectroscopy of aqueous amino acid solutions: Dynamics and interactions in aqueous glycine. J. Mol. Liq. 2005, 117, 93–98. [Google Scholar] [CrossRef]
- de Carvalho, M.F.; Mosquera, R.A.; Rivelino, R. A density functional theory study of the hydrogen bond interactions in glycine dimers. Chem. Phys. Lett. 2008, 445, 117–124. [Google Scholar] [CrossRef]
- Liszka, B.M.; Wagterveld, R.M.; Witkamp, G.J.; Otto, C. Calcium Carbonate Nucleation Investigated in a DoublePulse Experiment. Cryst. Growth Des. 2016, 16, 4839–4845. [Google Scholar] [CrossRef]
- Tsekova, D.; Popova, S.; Nanev, C. Nucleation rate determination by a concentration pulse technique: Application on ferritin crystals to show the effect of surface treatment of a substrate. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1588–1592. [Google Scholar] [CrossRef] [Green Version]
- Ibis, F.; Yu, T.W.; Penha, F.M.; Ganguly, D.; Nuhu, M.A.; van der Heijden, A.E.D.M.; Kramer, H.J.M.; Eral, H.B. Nucleation kinetics of calcium oxalate monohydrate as a function of pH, magnesium, and osteopontin concentration quantified with droplet microfluidics. Biomicrofluidics 2021, 15, 064103. [Google Scholar] [CrossRef]
- Selimovic, S.; Jia, Y.; Fraden, S. Measuring the Nucleation Rate of Lysozyme using Microfluidics. Cryst. Growth Des. 2009, 9, 1806–1810. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Zhang, M.; Liu, M.; Liu, Y.; Zhang, J.; Wang, K.; Tang, W.; Gong, J. Understanding Nucleation Mechanism of Mefenamic Acid: An Examination of Relation between Pre-assembly Structure in Solution and Nucleation Kinetics. Cryst. Growth Des. 2021, 21, 6473–6484. [Google Scholar] [CrossRef]
- Goh, L.; Chen, K.; Bhamidi, V.; He, G.; Kee, N.C.S.; Kenis, P.J.A.; Zukoski, I.C.F.; Braatz, R.D. A Stochastic Model for Nucleation Kinetics Determination in Droplet-Based Microfluidic Systems. Cryst. Growth Des. 2010, 10, 2515–2521. [Google Scholar] [CrossRef] [Green Version]
- Brandel, C.; ter Horst, J. Measuring induction times and crystal nucleation rates. Faraday. Discuss. 2015, 179, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Svärd, M.; Zeglinski, J.; Rasmuson, Å.C. Influence of Solvent and Solid-State Structure on Nucleation of Parabens. Cryst. Growth Des. 2014, 14, 3890–3902. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Rasmuson, Å.C. Nucleation of Butyl Paraben in Different Solvents. Cryst. Growth Des. 2013, 13, 4226–4238. [Google Scholar] [CrossRef]
- Sullivan, R.A.; Davey, R.J.; Sadiq, G.; Dent, G.; Back, K.R.; ter Horst, J.H.; Toroz, D.; Hammond, R.B. Revealing the Roles of Desolvation and Molecular Self-Assembly in Crystal Nucleation from Solution: Benzoic and p-Aminobenzoic Acids. Cryst. Growth Des. 2014, 14, 2689–2696. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Cabeza, A.J.; Davey, R.J.; Sachithananthan, S.S.; Smith, R.; Tang, S.K.; Vetter, T.; Xiao, Y. Aromatic stacking—A key step in nucleation. Chem. Commun. 2017, 53, 7905–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, H.; Kimura, Y.; Yamazaki, T.; Takigawa, I. Early Detection of Nucleation Events From Solution in LC-TEM by Machine Learning. Front. chem. 2022, 10, 818230. [Google Scholar] [CrossRef]
- Kimura, Y.; Katsuno, H.; Yamazaki, T. Possible embryos and precursors of crystalline nuclei of calcium carbonate observed by liquid-cell transmission electron microscopy. Faraday. Discuss. 2022. [Google Scholar] [CrossRef]
- Yao, L.; Ou, Z.; Luo, B.; Xu, C.; Chen, Q. Machine Learning to Reveal Nanoparticle Dynamics from Liquid-Phase TEM Videos. ACS Cent. Sci. 2020, 6, 1421–1430. [Google Scholar] [CrossRef]
- Khamar, D.; Zeglinski, J.; Mealey, D.; Rasmuson, A.C. Investigating the Role of Solvent–Solute Interaction in Crystal Nucleation of Salicylic Acid from Organic Solvents. J. Am. Chem. Soc. 2014, 136, 11664–11673. [Google Scholar] [CrossRef]
- Zheng, Z.X.; Hou, B.H.; Cheng, X.W.; Liu, W.Y.; Huang, X.; Bao, Y.; Wang, T.; Wang, Z.; Hao, H.X. The mechanism of solvent-mediated desolvation transformation of lenvatinib mesylate from dimethyl sulfoxide solvate to form D. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Xu, S.; Ma, Y.; Tang, W.; Zhang, F.; Wang, J.; Gong, J. Probing the Structural Pathway of Conformational Polymorph Nucleation by Comparing a Series of α,ω-Alkanedicarboxylic Acids. IUCrJ 2020, 7, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Chen, J.; Rosbottom, I.; Chen, W.; Guo, M.; Heng, J.Y.Y. Supersaturation and solvent dependent nucleation of carbamazepine polymorphs during rapid cooling crystallization. CrystEngComm 2021, 23, 813–823. [Google Scholar] [CrossRef]
- Tang, W.W.; Sima, A.D.; Gong, J.B.; Wang, J.K.; Li, T.L. Kinetic Difference between Concomitant Polymorphism and Solvent-Mediated Phase Transformation: A Case of Tolfenamic Acid. Cryst Growth Des. 2020, 20, 1779–1788. [Google Scholar] [CrossRef]
- Tang, W.W.; Quan, Y.F.; Gong, J.B.; Wang, J.K.; Yin, Q.X.; Li, T.L. Form selection of concomitant polymorphs: A case study informed by crystallization kinetics modeling. AIChE J. 2021, 67, e17129. [Google Scholar] [CrossRef]
- Rosbottom, I.; Ma, C.Y.; Turner, T.D.; O’Connell, R.A.; Loughrey, J.; Sadiq, G.; Davey, R.J.; Roberts, K.J. Influence of Solvent Composition on the Crystal Morphologyand Structure of p-Aminobenzoic Acid Crystallizedfrom Mixed Ethanol and Nitromethane Solutions. Cryst Growth Des. 2017, 17, 4151–4161. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R. Designer crystals: Intermolecular interactions, network structures and supramolecular synthons. Chem. Commun. 1997, 1475–1482. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen bonds and other intermolecular interactions in organometallic crystals. J. Chem. Soc. Dalton. 2000, 3745–3751. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: Solid state supramolecular synthesis. Curr. Opin. Solid State Mater. Sci. 1997, 2, 451–454. [Google Scholar] [CrossRef]
- Desiraju, P.G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Commins, P.; Dippenaar, A.B.; Li, L.; Hara, H.; Haynes, D.A.; Naumov, P. Mechanically compliant single crystals of a stable organic radical. Chem. Sci. 2021, 12, 6188–6193. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, J.; Zhang, Y.; Qin, M.; Wang, W. Nucleation process in the folding of a domain-swapped dimer. Phys. Rev. E 2010, 81, 021910. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Aihara, T.; Wakabayashi, K. Early nucleation events in the polymerization of actin, probed by time-resolved small-angle x-ray scattering. Sci. Rep. 2016, 6, 34539. [Google Scholar] [CrossRef]
- Mason, T.O.; Buell, A.K. The Kinetics, Thermodynamics and Mechanisms of Short Aromatic Peptide Self-Assembly. In Biological and Bio-Inspired Nanomaterials: Properties and Assembly Mechanisms; Perrett, S., Buell, A.K., Knowles, T.P.J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; Volume 1174, pp. 61–112. [Google Scholar]
- Liu, S.; Xu, S.; Tang, W.; Yu, B.; Hou, B.; Gong, J. Revealing the roles of solvation in D-mannitol’s polymorphic nucleation. CrystEngComm 2018, 20, 7435–7445. [Google Scholar] [CrossRef]
- Fours, B.; Cartigny, Y.; Petit, S.; Coquerel, G. Formation of new polymorphs without any nucleation step. Desolvation of the rimonabant monohydrate: Directional crystallisation concomitant to smooth dehydration. Faraday Discuss. 2015, 179, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Zong, S.; Wang, N.; Li, X.; Dong, Y.; Wang, T.; Huang, X.; Hao, H. Unveiling the self-association and desolvation in crystal nucleation. IUCrJ 2021, 8, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Huang, X.; Li, X.; Zong, S.; Wang, N.; Hao, H. Conformational Selectivity and Evolution Affected by the Desolvation Process. Cryst. Growth Des. 2022, 22, 1283–1291. [Google Scholar] [CrossRef]
- Liu, L.; Nakouzi, E.; Sushko, M.L.; Schenter, G.K.; Mundy, C.J.; Chun, J.; De Yoreo, J.J. Connecting energetics to dynamics in particle growth by oriented attachment using real-time observations. Nat. Commun. 2020, 11, 1045. [Google Scholar] [CrossRef]
- Du, W.; Yin, Q.X.; Bao, Y.; Xie, C.; Hou, B.H.; Hao, H.X.; Chen, W.; Wang, J.K.; Gong, J.B. Concomitant polymorphism of prasugrel hydrochloride in reactive crystallization. Ind. Eng. Chem. Res. 2013, 52, 16182–16189. [Google Scholar] [CrossRef]
- Bora, P.; Saikia, B.; Sarma, B. Oriented Crystallization on Organic Monolayers to Control Concomitant Polymorphism. Chem. -A Eur. J. 2020, 26, 699–710. [Google Scholar] [CrossRef]
- Suresh, M.; Srinivasan, K. Concomitant Polymorphism and Nucleation Control of DL -Methionine Through Antisolvent Crystallization. Chem. Eng. Technol. 2021, 44, 614–621. [Google Scholar] [CrossRef]
- Liu, J.; Shen, T.; Yang, Z.H.; Zhang, S.; Sun, G.Y. Multistep Heterogeneous Nucleation in Binary Mixtures of Charged Colloidal Spheres. J. Phys. Chem. Lett. 2017, 8, 4652–4658. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, S.; Jiao, F.; Newcomb, C.J.; Zhang, Y.; Prakash, A.; Liao, Z.; Baer, M.; Mundy, C.; Pfaendtner, J.; et al. Tuning crystallization pathways through sequence engineering of biomimetic polymers. Nat. Mater. 2017, 16, 767–774. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Y.; Yu, J. Qualitative discussion of prenucleation cluster role in crystallization of calcium carbonate under high concentration of magnesium based on experimental phenomena. J. Cryst. Growth 2017, 478, 77–84. [Google Scholar] [CrossRef]
- Li, S.; Tang, W.W.; Li, M.Y.; Wang, L.Y.; Yang, Y.; Gong, J.B. Understanding the Role of Citric Acid on the Crystallization Pathways of Calcium Oxalate Hydrates. Cryst Growth Des. 2019, 19, 3139–3147. [Google Scholar] [CrossRef]
- Finney, A.R.; Salvalaglio, M. Multiple pathways in NaCl homogeneous crystal nucleation. Faraday. Discuss. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lanaro, G.; Patey, G.N. Birth of NaCl Crystals: Insights from Molecular Simulations. J. Phys. Chem. B 2016, 120, 9076–9087. [Google Scholar] [CrossRef]
Figure 1.
Cluster formation and the associated changes of free energy during crystal nucleation. (a) Nanoscopical formation of new phase via molecular clustering and (b) free energy diagram of crystal nucleation showing the formation of critical nucleus size and supersaturation influence wherein a high supersaturation in the solution leads to a lower critical size of the nucleus (i.e., > ).
Figure 1.
Cluster formation and the associated changes of free energy during crystal nucleation. (a) Nanoscopical formation of new phase via molecular clustering and (b) free energy diagram of crystal nucleation showing the formation of critical nucleus size and supersaturation influence wherein a high supersaturation in the solution leads to a lower critical size of the nucleus (i.e., > ).

Figure 2.
(a) Schematic illustration of all possible changes in the size of a cluster of m molecules (solid lines) and the change in the size of critical cluster n* by the attachment and detachment of monomers only according to the Szilard–Farkas model. (b) Evolution of clusters toward the formation of a critical nucleus in the framework of CNT, The black line and red line indicate the process of monomer attachment and desorption respectively. Reprinted/adapted with permission from [13]. Copyright 2017, copyright Davey, R.J.; Schroeder, S.L.M.; Ter Horst, J.H. et al.
Figure 2.
(a) Schematic illustration of all possible changes in the size of a cluster of m molecules (solid lines) and the change in the size of critical cluster n* by the attachment and detachment of monomers only according to the Szilard–Farkas model. (b) Evolution of clusters toward the formation of a critical nucleus in the framework of CNT, The black line and red line indicate the process of monomer attachment and desorption respectively. Reprinted/adapted with permission from [13]. Copyright 2017, copyright Davey, R.J.; Schroeder, S.L.M.; Ter Horst, J.H. et al.

Figure 4.
Crystallization pathway of PNCs versus CNT. Reprinted/adapted with permission from [21]. Copyright 2017, copyright Gebauer, D. et al.
Figure 4.
Crystallization pathway of PNCs versus CNT. Reprinted/adapted with permission from [21]. Copyright 2017, copyright Gebauer, D. et al.

Figure 5.
Schematic representation of solvent-dependent self-association and polymorphic crystallization.
Figure 5.
Schematic representation of solvent-dependent self-association and polymorphic crystallization.

Figure 6.
(a–c) Monomeric and dimeric structure model of TFA with selected inter-proton NOE labeled. (d,e) 2D NOESY spectra of TFA in ethanol-d6 at 298 K. Positive and negative signals are represented by blue and red contours, respectively [108]. Reprinted/adapted with permission from [108]. Copyright 2017, copyright Tang, W.; Mo, H.; Zhang, M. et al.
Figure 6.
(a–c) Monomeric and dimeric structure model of TFA with selected inter-proton NOE labeled. (d,e) 2D NOESY spectra of TFA in ethanol-d6 at 298 K. Positive and negative signals are represented by blue and red contours, respectively [108]. Reprinted/adapted with permission from [108]. Copyright 2017, copyright Tang, W.; Mo, H.; Zhang, M. et al.

Figure 7.
Hydrogen-bonded cyclic dimer (a) and open-chain dimer (b) respectively presented in α form and γ form of glycine [113]. Reprinted/adapted with permission from [113]. Copyright 2012, copyright Yani, Y.; Chow, P.S.; Tan, R.B.H.

Figure 8.
Proposed nucleation pathway of concomitant nucleation of a dipolymorphic system. The colors of solvated monomers indicate different solvation states [136]. Reprinted/adapted with permission from [136]. Copyright 2020, copyright Tang, W.; Sima, A.D.; Gong, J. et al.

Figure 9.
Hierarchical self−assembly of benzoic acid molecules in solution dictated by multiple intermolecular interactions: (a) higher−order self−assembly pathways in the pre−nucleation stage. (b) Electrostatic potential of benzoic acid molecules with hydrogen−bonding interactions. (c) Hydrogen−bonded dimer motif. (d) Nucleophilic and electrophilic Fukui function mapped on the van der Waals surface. (e) Hierarchical tetramer motif [35]. Reprinted/adapted with permission from [35]. Copyright 2017, copyright Tang, W.; Zhang, M.; Mo, H.; et al.
Figure 9.
Hierarchical self−assembly of benzoic acid molecules in solution dictated by multiple intermolecular interactions: (a) higher−order self−assembly pathways in the pre−nucleation stage. (b) Electrostatic potential of benzoic acid molecules with hydrogen−bonding interactions. (c) Hydrogen−bonded dimer motif. (d) Nucleophilic and electrophilic Fukui function mapped on the van der Waals surface. (e) Hierarchical tetramer motif [35]. Reprinted/adapted with permission from [35]. Copyright 2017, copyright Tang, W.; Zhang, M.; Mo, H.; et al.

Figure 10.
Schematic illustration of molecular aggregation-based multiple nucleation pathways: various pathways of evolutions of density and structure order of solutes’ associates, aggregates, or clusters accompanying the change of free energy in different solution systems. Pathway Ⅰ represents the self-assembly and nucleation pathway of benzoic acid molecules in toluene; pathway Ⅱ denotes the scenario of crystal nucleation of TFA in toluene, and pathway Ⅲ is the case of gly in water and other systems wherein (solvated) monomers are dominant in solution. The blue dashed line is the pathway of two-step nucleation. The light grey dash line represents the nucleation pathway of CNT.
Figure 10.
Schematic illustration of molecular aggregation-based multiple nucleation pathways: various pathways of evolutions of density and structure order of solutes’ associates, aggregates, or clusters accompanying the change of free energy in different solution systems. Pathway Ⅰ represents the self-assembly and nucleation pathway of benzoic acid molecules in toluene; pathway Ⅱ denotes the scenario of crystal nucleation of TFA in toluene, and pathway Ⅲ is the case of gly in water and other systems wherein (solvated) monomers are dominant in solution. The blue dashed line is the pathway of two-step nucleation. The light grey dash line represents the nucleation pathway of CNT.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhou, J.; Zhou, Y.; Tang, W. Molecular Mechanism of Organic Crystal Nucleation: A Perspective of Solution Chemistry and Polymorphism. Crystals 2022, 12, 980. https://doi.org/10.3390/cryst12070980
AMA Style
Zhou J, Zhou Y, Tang W. Molecular Mechanism of Organic Crystal Nucleation: A Perspective of Solution Chemistry and Polymorphism. Crystals. 2022; 12(7):980. https://doi.org/10.3390/cryst12070980
Chicago/Turabian StyleZhou, Jianmin, Yixin Zhou, and Weiwei Tang. 2022. "Molecular Mechanism of Organic Crystal Nucleation: A Perspective of Solution Chemistry and Polymorphism" Crystals 12, no. 7: 980. https://doi.org/10.3390/cryst12070980
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.