
Salivarian Trypanosomes Have Adopted Intricate Host-Pathogen Interaction Mechanisms That Ensure Survival in Plain Sight of the Adaptive Immune System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
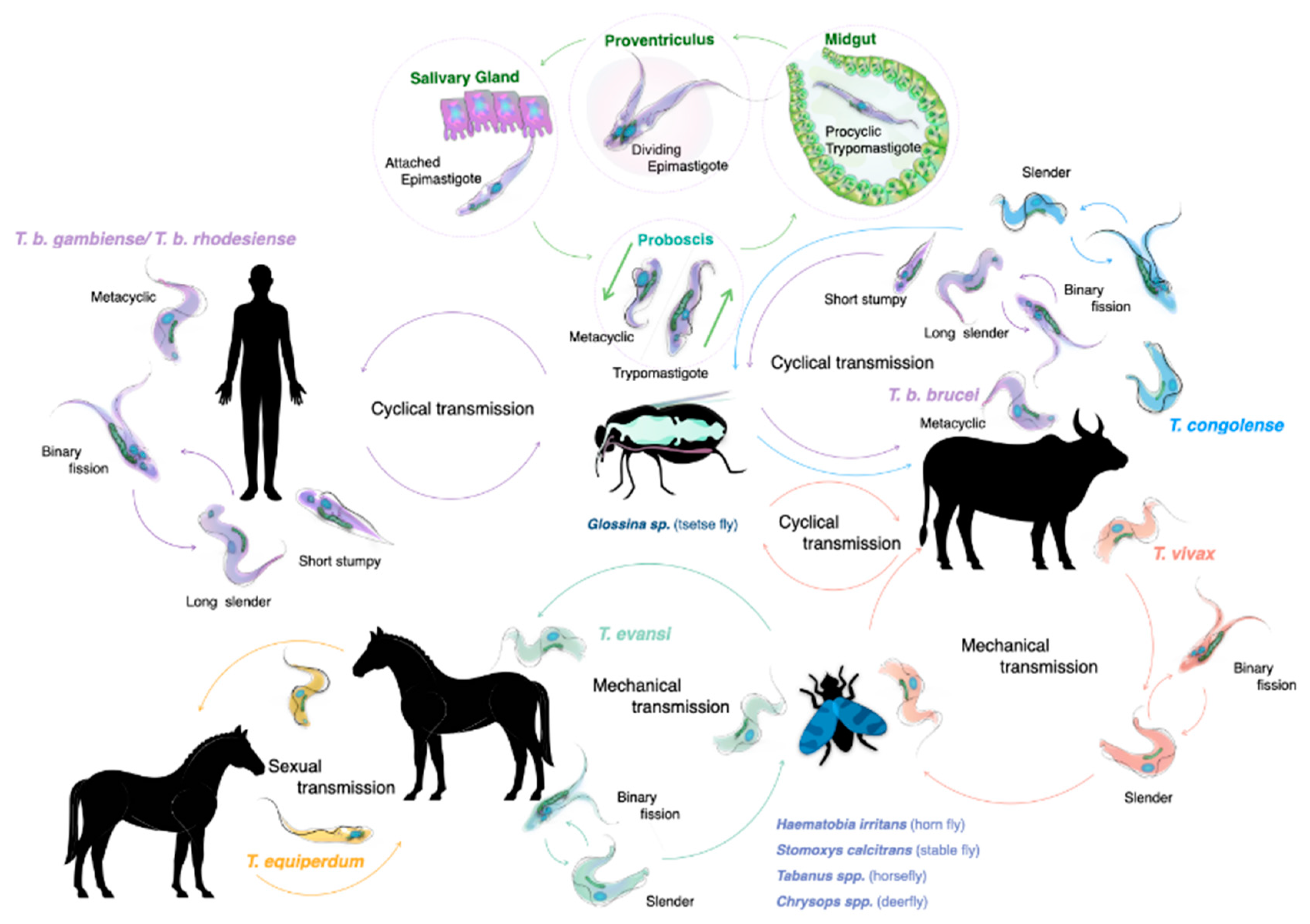
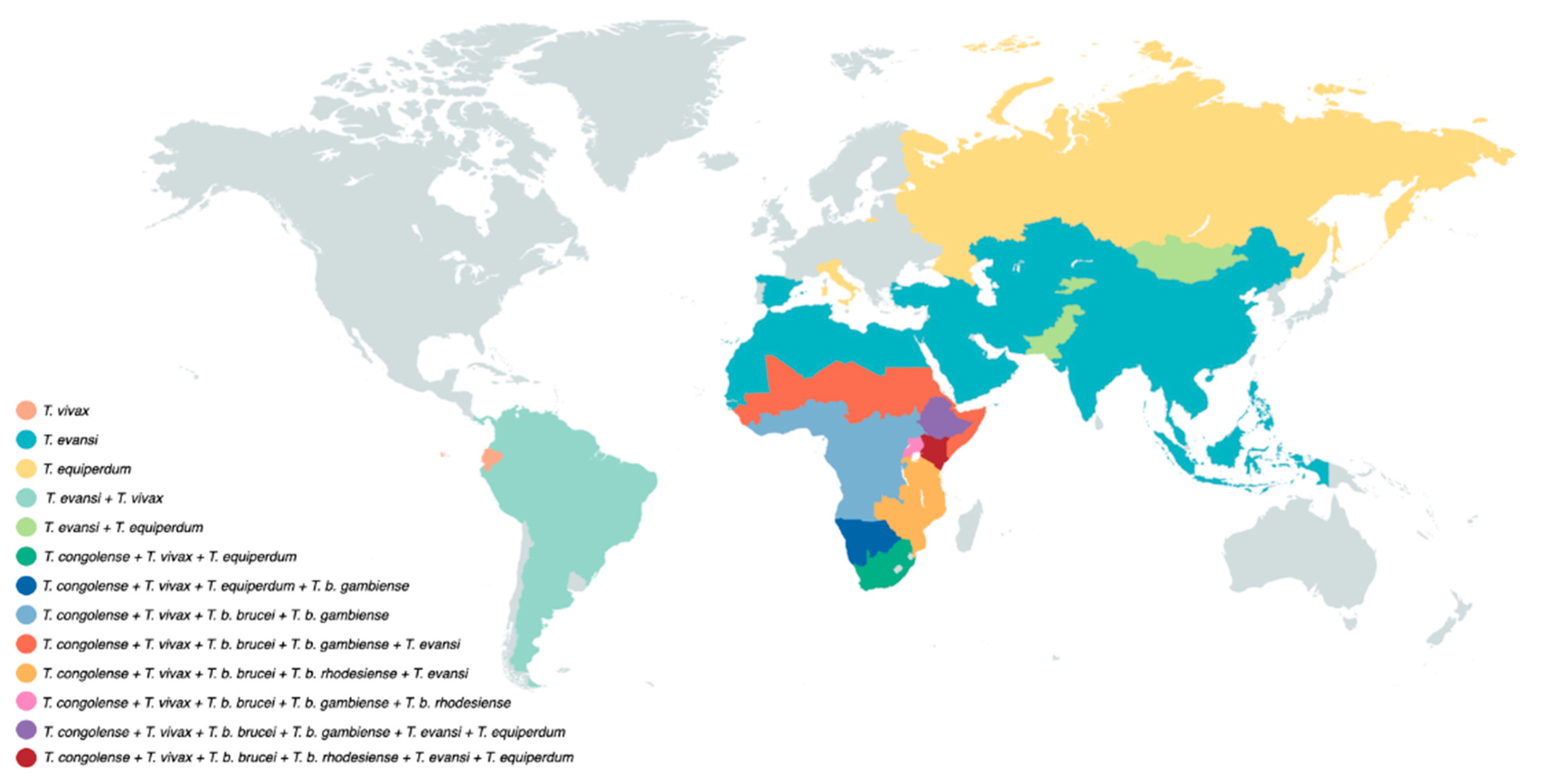
2. The Life Cycle of Trypanosomes
3. Trypanosomiasis and the Human Biochemical Defense System
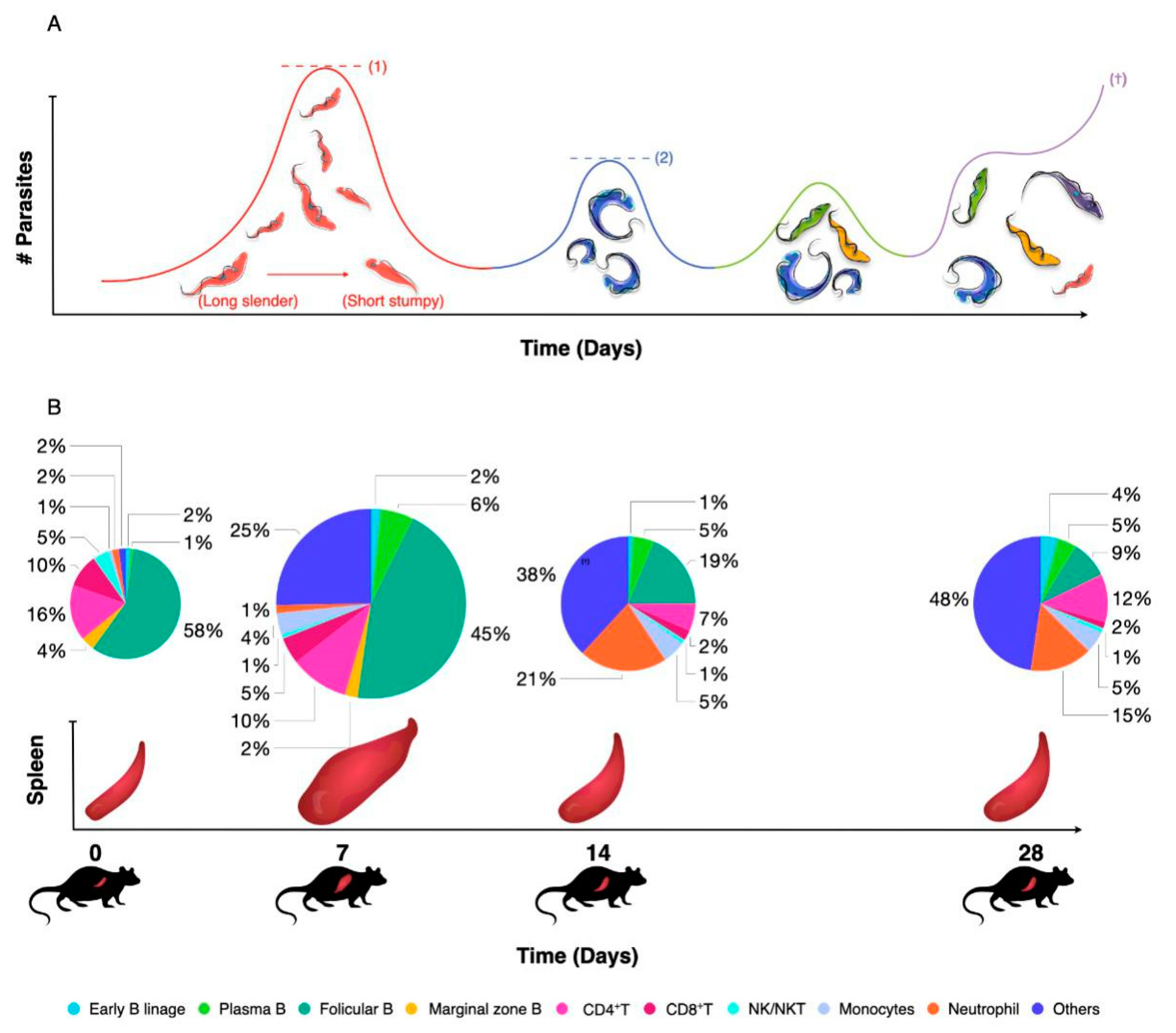
4. Innate and Adaptive Immunity to Trypanosomiasis
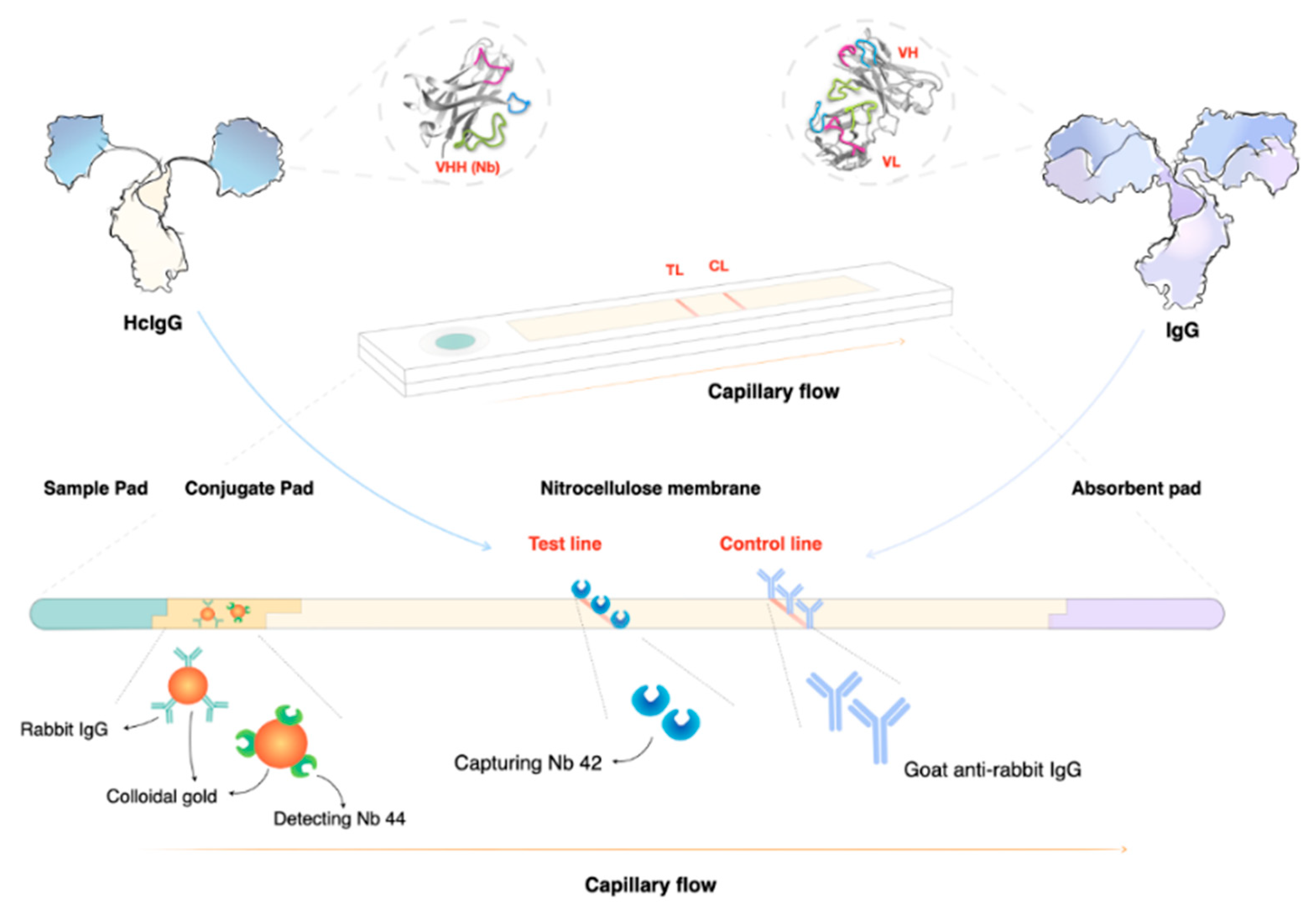
5. Recent Advances in the Diagnosis of HAT and AT
6. Recent Advances in Treatment of HAT
7. The Lack of Anti-Trypanosome Vaccination Still Hampers Sustainable Disease Control
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hutchinson, O.C.; Fèvre, E.M.; Carrington, M.; Welburn, S.C. Lessons learned from the emergence of a new Trypanosoma brucei rhodesiense sleeping sickness focus in Uganda. Lancet Infect. Dis. 2003, 3, 42–45. [Google Scholar] [CrossRef]
- Squarre, D.; Hayashida, K.; Gaithuma, A.; Chambaro, H.; Kawai, N.; Moonga, L.; Namangala, B.; Sugimoto, C.; Yamagishi, J. Diversity of trypanosomes in wildlife of the Kafue ecosystem, Zambia. Int. J. Parasitol. Parasites Wildl. 2020, 12, 34–41. [Google Scholar] [CrossRef]
- Moloo, S.K.; Grootenhuis, J.G.; Jenni, L.; Brun, R.; van Meirvenne, N.; Murray, M. Trypanosoma brucei rhodesiense: Variation in human serum resistance after transmission between bushbuck and domestic ruminants by Glossina morsitans morsitans. Acta Trop. 1995, 59, 255–258. [Google Scholar] [CrossRef]
- Franco, J.R.; Cecchi, G.; Priotto, G.; Paone, M.; Diarra, A.; Grout, L.; Simarro, P.P.; Zhao, W.; Argaw, D. Monitoring the elimination of human African trypanosomiasis at continental and country level: Update to 2018. PLoS Negl. Trop. Dis. 2020, 14, e0008261. [Google Scholar] [CrossRef] [PubMed]
- Jamonneau, V.; Truc, P.; Grebaut, P.; Herder, S.; Ravel, S.; Solano, P.; De Meeus, T. Trypanosoma brucei gambiense Group 2: The Unusual Suspect. Trends Parasitol. 2019, 35, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Aregawi, W.G.; Agga, G.E.; Abdi, R.D.; Buscher, P. Systematic review and meta-analysis on the global distribution, host range, and prevalence of Trypanosoma evansi. Parasites Vectors 2019, 12, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaro, M.; Munyard, K.A.; Stear, M.J.; Groth, D.M. Combatting African Animal Trypanosomiasis (AAT) in livestock: The potential role of trypanotolerance. Vet. Parasitol. 2016, 225, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Radwanska, M.; Vereecke, N.; Deleeuw, V.; Pinto, J.; Magez, S. Salivarian Trypanosomosis: A Review of Parasites Involved, Their Global Distribution and Their Interaction with the Innate and Adaptive Mammalian Host Immune System. Front. Immunol. 2018, 9, 2253. [Google Scholar] [CrossRef]
- Kennedy, P.G.E.; Rodgers, J. Clinical and Neuropathogenetic Aspects of Human African Trypanosomiasis. Front. Immunol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W. The origins of the trypanosome genome strains Trypanosoma brucei brucei TREU 927, T. b. gambiense DAL 972, T. vivax Y486 and T. congolense IL3000. Parasites Vectors 2012, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Magez, S.; Pinto Torres, J.E.; Obishakin, E.; Radwanska, M. Infections with Extracellular Trypanosomes Require Control by Efficient Innate Immune Mechanisms and Can Result in the Destruction of the Mammalian Humoral Immune System. Front. Immunol. 2020, 11, 382. [Google Scholar] [CrossRef] [Green Version]
- Muhanguzi, D.; Picozzi, K.; Hatendorf, J.; Thrusfield, M.; Welburn, S.C.; Kabasa, J.D.; Waiswa, C. Improvements on restricted insecticide application protocol for control of Human and Animal African Trypanosomiasis in eastern Uganda. PLoS Negl. Trop. Dis. 2014, 8, e3284. [Google Scholar] [CrossRef] [Green Version]
- Lehane, M.; Alfaroukh, I.; Bucheton, B.; Camara, M.; Harris, A.; Kaba, D.; Lumbala, C.; Peka, M.; Rayaisse, J.B.; Waiswa, C.; et al. Tsetse Control and the Elimination of Gambian Sleeping Sickness. PLoS Negl. Trop. Dis. 2016, 10, e0004437. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.G.; My, L.N.; Dung, T.V.; Thanh, N.G.; Tam, P.T.; Vercruysse, J.; Goddeeris, B.M. The influence of T. evansi infection on the immuno-responsiveness of experimentally infected water buffaloes. Vet. Parasitol. 2001, 102, 225–234. [Google Scholar] [CrossRef]
- Wamwiri, F.N.; Changasi, R.E. Tsetse Flies (Glossina) as Vectors of Human African Trypanosomiasis: A Review. Biomed. Res. Int. 2016, 2016, 6201350. [Google Scholar] [CrossRef] [Green Version]
- Van Den Abbeele, J.; Claes, Y.; van Bockstaele, D.; Le Ray, D.; Coosemans, M. Trypanosoma brucei spp. development in the tsetse fly: Characterization of the post-mesocyclic stages in the foregut and proboscis. Parasitology 1999, 118 Pt 5, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W. Liaisons dangereuses: Sexual recombination among pathogenic trypanosomes. Res. Microbiol. 2015, 166, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W.; Peacock, L. Fluorescent proteins reveal what trypanosomes get up to inside the tsetse fly. Parasites Vectors 2019, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Gluenz, E.; Peacock, L.; Gibson, W.; Gull, K.; Carrington, M. The heart of darkness: Growth and form of Trypanosoma brucei in the tsetse fly. Trends Parasitol. 2009, 25, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Abbeele, J.; Caljon, G.; De Ridder, K.; De Baetselier, P.; Coosemans, M. Trypanosoma brucei modifies the tsetse salivary composition, altering the fly feeding behavior that favors parasite transmission. PLoS Pathog. 2010, 6, e1000926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caljon, G.; Van Den Abbeele, J.; Stijlemans, B.; Coosemans, M.; De Baetselier, P.; Magez, S. Tsetse fly saliva accelerates the onset of Trypanosoma brucei infection in a mouse model associated with a reduced host inflammatory response. Infect. Immun. 2006, 74, 6324–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caljon, G.; De Ridder, K.; De Baetselier, P.; Coosemans, M.; Van Den Abbeele, J. Identification of a tsetse fly salivary protein with dual inhibitory action on human platelet aggregation. PLoS ONE 2010, 5, e9671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolev, N.G.; Ramsdell, T.K.; Tschudi, C. Temperature shift activates bloodstream VSG expression site promoters in Trypanosoma brucei. Mol. Biochem. Parasitol. 2018, 226, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Bangs, J.D. Evolution of Antigenic Variation in African Trypanosomes: Variant Surface Glycoprotein Expression, Structure, and Function. Bioessays 2018, 40, e1800181. [Google Scholar] [CrossRef] [PubMed]
- Vassella, E.; Reuner, B.; Yutzy, B.; Boshart, M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 1997, 110 Pt 21, 2661–2671. [Google Scholar] [CrossRef]
- Rojas, F.; Silvester, E.; Young, J.; Milne, R.; Tettey, M.; Houston, D.R.; Walkinshaw, M.D.; Perez-Pi, I.; Auer, M.; Denton, H.; et al. Oligopeptide Signaling through TbGPR89 Drives Trypanosome Quorum Sensing. Cell 2019, 176, 306–317.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, F.; Matthews, K.R. Quorum sensing in African trypanosomes. Curr. Opin. Microbiol. 2019, 52, 124–129. [Google Scholar] [CrossRef]
- Magez, S.; Truyens, C.; Merimi, M.; Radwanska, M.; Stijlemans, B.; Brouckaert, P.; Brombacher, F.; Pays, E.; De Baetselier, P. P75 tumor necrosis factor-receptor shedding occurs as a protective host response during African trypanosomiasis. J. Infect. Dis. 2004, 189, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Magez, S.; Schwegmann, A.; Atkinson, R.; Claes, F.; Drennan, M.; De Baetselier, P.; Brombacher, F. The role of B-cells and IgM antibodies in parasitemia, anemia, and VSG switching in Trypanosoma brucei-infected mice. PLoS Pathog. 2008, 4, e1000122. [Google Scholar] [CrossRef] [Green Version]
- Peacock, L.; Cook, S.; Ferris, V.; Bailey, M.; Gibson, W. The life cycle of Trypanosoma (Nannomonas) congolense in the tsetse fly. Parasites Vectors 2012, 5, 109. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W.; Peacock, L.; Hutchinson, R. Microarchitecture of the tsetse fly proboscis. Parasites Vectors 2017, 10, 430. [Google Scholar] [CrossRef] [Green Version]
- Ooi, C.P.; Schuster, S.; Cren-Travaillé, C.; Bertiaux, E.; Cosson, A.; Goyard, S.; Perrot, S.; Rotureau, B. The Cyclical Development of Trypanosoma vivax in the Tsetse Fly Involves an Asymmetric Division. Front. Cell Infect. Microbiol. 2016, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.J.; Tweedie, A.; Black, A.; Pinchbeck, G.L.; Christley, R.M.; Schoenefeld, A.; Hertz-Fowler, C.; MacLeod, A.; Turner, C.M.; Tait, A. Discovery of mating in the major African livestock pathogen Trypanosoma congolense. PLoS ONE 2009, 4, e5564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, P.; Fase-Fowler, F.; Gibson, W.C. Kinetoplast DNA of Trypanosoma evansi. Mol. Biochem. Parasitol. 1987, 23, 31–38. [Google Scholar] [CrossRef]
- Zweygarth, E.; Kaminsky, R.; Webster, P. Trypanosoma brucei evansi: Dyskinetoplasia and loss of infectivity after long-term in vitro cultivation. Acta Trop. 1990, 48, 95–99. [Google Scholar] [CrossRef]
- Desquesnes, M.; Dargantes, A.; Lai, D.H.; Lun, Z.R.; Holzmuller, P.; Jittapalapong, S. Trypanosoma evansi and surra: A review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed. Res. Int. 2013, 2013, 321237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desquesnes, M.; Holzmuller, P.; Lai, D.H.; Dargantes, A.; Lun, Z.R.; Jittaplapong, S. Trypanosoma evansi and surra: A review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, and pathogenic effects. Biomed. Res. Int. 2013, 2013, 194176. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, C.; Desquesnes, M.; Touratier, L.; Buscher, P. Trypanosoma evansi: Recent outbreaks in Europe. Vet. Parasitol. 2010, 174, 26–29. [Google Scholar] [CrossRef]
- Gutierrez, C.; Tamarit, A.; Gonzalez-Martin, M.; Tejedor-Junco, M.T. Control and eventual eradication of Trypanosoma evansi infection in dromedary camels after an episodic outbreak in mainland Spain: An example in a non-endemic area. Vet. Parasitol. 2014, 204, 153–157. [Google Scholar] [CrossRef]
- Molinari, J.; Moreno, S.A. Trypanosoma brucei Plimmer & Bradford, 1899 is a synonym of T. evansi (Steel, 1885) according to current knowledge and by application of nomenclature rules. Syst. Parasitol. 2018, 95, 249–256. [Google Scholar] [CrossRef]
- WHO. Human African Trypanosomiasis: Epidemiological Situation. Available online: https://www.who.int/trypanosomiasis_african/country/en/ (accessed on 14 May 2021).
- Kennedy, P.G.E. Update on human African trypanosomiasis (sleeping sickness). J. Neurol. 2019, 266, 2334–2337. [Google Scholar] [CrossRef] [Green Version]
- Masocha, W.; Kristensson, K. Human African trypanosomiasis: How do the parasites enter and cause dysfunctions of the nervous system in murine models? Brain Res. Bull. 2019, 145, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Bentivoglio, M.; Kristensson, K.; Rottenberg, M.E. Circumventricular Organs and Parasite Neurotropism: Neglected Gates to the Brain? Front. Immunol. 2018, 9, 2877. [Google Scholar] [CrossRef] [PubMed]
- Raper, J.; Fung, R.; Ghiso, J.; Nussenzweig, V.; Tomlinson, S. Characterization of a novel trypanosome lytic factor from human serum. Infect. Immun. 1999, 67, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.; Genovese, G.; Canon, C.; Kovacsics, D.; Higgins, M.K.; Carrington, M.; Winkler, C.A.; Kopp, J.; Rotimi, C.; Adeyemo, A.; et al. Evolution of the primate trypanolytic factor APOL1. Proc. Natl. Acad. Sci. USA 2014, 111, E2130–E2139. [Google Scholar] [CrossRef] [Green Version]
- Vanhollebeke, B.; Nielsen, M.J.; Watanabe, Y.; Truc, P.; Vanhamme, L.; Nakajima, K.; Moestrup, S.K.; Pays, E. Distinct roles of haptoglobin-related protein and apolipoprotein L-I in trypanolysis by human serum. Proc. Natl. Acad. Sci. USA 2007, 104, 4118–4123. [Google Scholar] [CrossRef] [Green Version]
- Vanwalleghem, G.; Fontaine, F.; Lecordier, L.; Tebabi, P.; Klewe, K.; Nolan, D.P.; Yamaryo-Botte, Y.; Botte, C.; Kremer, A.; Burkard, G.S.; et al. Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat. Commun. 2015, 6, 8078. [Google Scholar] [CrossRef] [Green Version]
- Thomson, R.; Finkelstein, A. Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: Relevance to trypanosome lysis. Proc. Natl. Acad. Sci. USA 2015, 112, 2894–2899. [Google Scholar] [CrossRef] [Green Version]
- Verdi, J.; Zipkin, R.; Hillman, E.; Gertsch, R.A.; Pangburn, S.J.; Thomson, R.; Papavasiliou, N.; Sternberg, J.; Raper, J. Inducible Germline IgMs Bridge Trypanosome Lytic Factor Assembly and Parasite Recognition. Cell Host Microbe 2020, 28, 79–88.e74. [Google Scholar] [CrossRef]
- Raper, J.; Nussenzweig, V.; Tomlinson, S. The main lytic factor of Trypanosoma brucei brucei in normal human serum is not high density lipoprotein. J. Exp. Med. 1996, 183, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina Portela, M.P.; Raper, J.; Tomlinson, S. An investigation into the mechanism of trypanosome lysis by human serum factors. Mol. Biochem. Parasitol. 2000, 110, 273–282. [Google Scholar] [CrossRef]
- Salmon, D.; Hanocq-Quertier, J.; Paturiaux-Hanocq, F.; Pays, A.; Tebabi, P.; Nolan, D.P.; Michel, A.; Pays, E. Characterization of the ligand-binding site of the transferrin receptor in Trypanosoma brucei demonstrates a structural relationship with the N-terminal domain of the variant surface glycoprotein. EMBO J. 1997, 16, 7272–7278. [Google Scholar] [CrossRef] [PubMed]
- Kariuki, C.K.; Stijlemans, B.; Magez, S. The Trypanosomal Transferrin Receptor of Trypanosoma brucei—A Review. Trop. Med. Infect. Dis. 2019, 4, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhollebeke, B.; De Muylder, G.; Nielsen, M.J.; Pays, A.; Tebabi, P.; Dieu, M.; Raes, M.; Moestrup, S.K.; Pays, E. A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science 2008, 320, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Bullard, W.; Kieft, R.; Capewell, P.; Veitch, N.J.; Macleod, A.; Hajduk, S.L. Haptoglobin-hemoglobin receptor independent killing of African trypanosomes by human serum and trypanosome lytic factors. Virulence 2012, 3, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecordier, L.; Uzureau, P.; Tebabi, P.; Perez-Morga, D.; Nolan, D.; Schumann Burkard, G.; Roditi, I.; Pays, E. Identification of Trypanosoma brucei components involved in trypanolysis by normal human serum. Mol. Microbiol. 2014, 94, 625–636. [Google Scholar] [CrossRef]
- Vanhamme, L.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Nolan, D.P.; Lins, L.; Van Den Abbeele, J.; Pays, A.; Tebabi, P.; Van Xong, H.; Jacquet, A.; et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 2003, 422, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N.; Magez, S.; Stijlemans, B.; Conrath, K.; Vanhollebeke, B.; Pays, E.; Muyldermans, S.; De Baetselier, P. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat. Med. 2006, 12, 580–584. [Google Scholar] [CrossRef]
- Cooper, A.; Capewell, P.; Clucas, C.; Veitch, N.; Weir, W.; Thomson, R.; Raper, J.; MacLeod, A. A Primate APOL1 Variant That Kills Trypanosoma brucei gambiense. PLoS Negl. Trop. Dis. 2016, 10, e0004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, R.; Molina-Portela, P.; Mott, H.; Carrington, M.; Raper, J. Hydrodynamic gene delivery of baboon trypanosome lytic factor eliminates both animal and human-infective African trypanosomes. Proc. Natl. Acad. Sci. USA 2009, 106, 19509–19514. [Google Scholar] [CrossRef] [Green Version]
- De Greef, C.; Imberechts, H.; Matthyssens, G.; Van Meirvenne, N.; Hamers, R. A gene expressed only in serum-resistant variants of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 1989, 36, 169–176. [Google Scholar] [CrossRef]
- De Greef, C.; Chimfwembe, E.; Kihang’a Wabacha, J.; Bajyana Songa, E.; Hamers, R. Only the serum-resistant bloodstream forms of Trypanosoma brucei rhodesiense express the serum resistance associated (SRA) protein. Ann. Soc. Belg. Med. Trop. 1992, 72 (Suppl. S1), 13–21. [Google Scholar]
- De Greef, C.; Hamers, R. The serum resistance-associated (SRA) gene of Trypanosoma brucei rhodesiense encodes a variant surface glycoprotein-like protein. Mol. Biochem. Parasitol. 1994, 68, 277–284. [Google Scholar] [CrossRef]
- Van Xong, H.; Vanhamme, L.; Chamekh, M.; Chimfwembe, C.E.; Van Den Abbeele, J.; Pays, A.; Van Meirvenne, N.; Hamers, R.; De Baetselier, P.; Pays, E. A VSG Expression Site-Associated Gene Confers Resistance to Human Serum in Trypanosoma rhodesiense. Cell 1998, 95, 839–846. [Google Scholar] [CrossRef] [Green Version]
- Radwanska, M.; Claes, F.; Magez, S.; Magnus, E.; Perez-Morga, D.; Pays, E.; Buscher, P. Novel primer sequences for polymerase chain reaction-based detection of Trypanosoma brucei gambiense. Am. J. Trop. Med. Hyg. 2002, 67, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Kieft, R.; Capewell, P.; Turner, C.M.; Veitch, N.J.; MacLeod, A.; Hajduk, S. Mechanism of Trypanosoma brucei gambiense (group 1) resistance to human trypanosome lytic factor. Proc. Natl. Acad. Sci. USA 2010, 107, 16137–16141. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.K.; Tkachenko, O.; Brown, A.; Reed, J.; Raper, J.; Carrington, M. Structure of the trypanosome haptoglobin-hemoglobin receptor and implications for nutrient uptake and innate immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 1905–1910. [Google Scholar] [CrossRef] [Green Version]
- DeJesus, E.; Kieft, R.; Albright, B.; Stephens, N.A.; Hajduk, S.L. A single amino acid substitution in the group 1 Trypanosoma brucei gambiense haptoglobin-hemoglobin receptor abolishes TLF-1 binding. PLoS Pathog. 2013, 9, e1003317. [Google Scholar] [CrossRef] [Green Version]
- Lane-Serff, H.; MacGregor, P.; Lowe, E.D.; Carrington, M.; Higgins, M.K. Structural basis for ligand and innate immunity factor uptake by the trypanosome haptoglobin-haemoglobin receptor. eLife 2014, 3, e05553. [Google Scholar] [CrossRef] [PubMed]
- Capewell, P.; Clucas, C.; DeJesus, E.; Kieft, R.; Hajduk, S.; Veitch, N.; Steketee, P.C.; Cooper, A.; Weir, W.; MacLeod, A. The TgsGP gene is essential for resistance to human serum in Trypanosoma brucei gambiense. PLoS Pathog. 2013, 9, e1003686. [Google Scholar] [CrossRef] [Green Version]
- Uzureau, P.; Uzureau, S.; Lecordier, L.; Fontaine, F.; Tebabi, P.; Homble, F.; Grelard, A.; Zhendre, V.; Nolan, D.P.; Lins, L.; et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature 2013, 501, 430–434. [Google Scholar] [CrossRef]
- Capewell, P.; Veitch, N.J.; Turner, C.M.; Raper, J.; Berriman, M.; Hajduk, S.L.; MacLeod, A. Differences between Trypanosoma brucei gambiense groups 1 and 2 in their resistance to killing by trypanolytic factor 1. PLoS Negl. Trop. Dis. 2011, 5, e1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capewell, P.; Cooper, A.; Clucas, C.; Weir, W.; Macleod, A. A co-evolutionary arms race: Trypanosomes shaping the human genome, humans shaping the trypanosome genome. Parasitology 2015, 142 (Suppl. S1), S108–S119. [Google Scholar] [CrossRef] [Green Version]
- Truc, P.; Büscher, P.; Cuny, G.; Gonzatti, M.I.; Jannin, J.; Joshi, P.; Juyal, P.; Lun, Z.R.; Mattioli, R.; Pays, E.; et al. Atypical human infections by animal trypanosomes. PLoS Negl. Trop. Dis. 2013, 7, e2256. [Google Scholar] [CrossRef] [Green Version]
- Vanhollebeke, B.; Truc, P.; Poelvoorde, P.; Pays, A.; Joshi, P.P.; Katti, R.; Jannin, J.G.; Pays, E. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N. Eng. J. Med. 2006, 355, 2752–2756. [Google Scholar] [CrossRef]
- Van Vinh Chau, N.; Buu Chau, L.; Desquesnes, M.; Herder, S.; Phu Huong Lan, N.; Campbell, J.I.; Van Cuong, N.; Yimming, B.; Chalermwong, P.; Jittapalapong, S.; et al. A Clinical and Epidemiological Investigation of the First Reported Human Infection with the Zoonotic Parasite Trypanosoma evansi in Southeast Asia. Clin. Infect. Dis. 2016, 62, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Kamidi, C.M.; Saarman, N.P.; Dion, K.; Mireji, P.O.; Ouma, C.; Murilla, G.; Aksoy, S.; Schnaufer, A.; Caccone, A. Multiple evolutionary origins of Trypanosoma evansi in Kenya. PLoS Negl. Trop. Dis. 2017, 11, e0005895. [Google Scholar] [CrossRef] [Green Version]
- Carnes, J.; Anupama, A.; Balmer, O.; Jackson, A.; Lewis, M.; Brown, R.; Cestari, I.; Desquesnes, M.; Gendrin, C.; Hertz-Fowler, C.; et al. Genome and phylogenetic analyses of Trypanosoma evansi reveal extensive similarity to T. brucei and multiple independent origins for dyskinetoplasty. PLoS Negl. Trop. Dis. 2015, 9, e3404. [Google Scholar] [CrossRef] [Green Version]
- Engstler, M.; Pfohl, T.; Herminghaus, S.; Boshart, M.; Wiegertjes, G.; Heddergott, N.; Overath, P. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell 2007, 131, 505–515. [Google Scholar] [CrossRef] [Green Version]
- La Greca, F.; Haynes, C.; Stijlemans, B.; De Trez, C.; Magez, S. Antibody-mediated control of Trypanosoma vivax infection fails in the absence of tumour necrosis factor. Parasite Immunol. 2014, 36, 271–276. [Google Scholar] [CrossRef]
- Frevert, U.; Reinwald, E. Trypanosoma congolense bloodstream forms evade complement lysis in vitro by shedding of immune complexes. Eur. J. Cell Biol. 1990, 52, 264–269. [Google Scholar]
- Magez, S.; Stijlemans, B.; Radwanska, M.; Pays, E.; Ferguson, M.A.; De Baetselier, P. The glycosyl-inositol-phosphate and dimyristoylglycerol moieties of the glycosylphosphatidylinositol anchor of the trypanosome variant-specific surface glycoprotein are distinct macrophage-activating factors. J. Immunol. 1998, 160, 1949–1956. [Google Scholar]
- Hertz, C.J.; Filutowicz, H.; Mansfield, J.M. Resistance to the African trypanosomes is IFN-gamma dependent. J. Immunol. 1998, 161, 6775–6783. [Google Scholar]
- Wu, H.; Liu, G.; Shi, M. Interferon Gamma in African Trypanosome Infections: Friends or Foes? Front. Immunol. 2017, 8, 1105. [Google Scholar] [CrossRef]
- Magez, S.; Lucas, R.; Darji, A.; Songa, E.B.; Hamers, R.; De Baetselier, P. Murine tumour necrosis factor plays a protective role during the initial phase of the experimental infection with Trypanosoma brucei brucei. Parasite Immunol. 1993, 15, 635–641. [Google Scholar] [CrossRef]
- Deleeuw, V.; Pham, H.T.T.; De Poorter, I.; Janssens, I.; De Trez, C.; Radwanska, M.; Magez, S. Trypanosoma brucei brucei causes a rapid and persistent influx of neutrophils in the spleen of infected mice. Parasite Immunol. 2019, 41, e12664. [Google Scholar] [CrossRef] [Green Version]
- De Trez, C.; Stijlemans, B.; Bockstal, V.; Cnops, J.; Korf, H.; Van Snick, J.; Caljon, G.; Muraille, E.; Humphreys, I.R.; Boon, L.; et al. A Critical Blimp-1-Dependent IL-10 Regulatory Pathway in T Cells Protects from a Lethal Pro-inflammatory Cytokine Storm During Acute Experimental Trypanosoma brucei Infection. Front. Immunol. 2020, 11, 1085. [Google Scholar] [CrossRef]
- Cnops, J.; De Trez, C.; Stijlemans, B.; Keirsse, J.; Kauffmann, F.; Barkhuizen, M.; Keeton, R.; Boon, L.; Brombacher, F.; Magez, S. NK-, NKT- and CD8-Derived IFNgamma Drives Myeloid Cell Activation and Erythrophagocytosis, Resulting in Trypanosomosis-Associated Acute Anemia. PLoS Pathog. 2015, 11, e1004964. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Ogunremi, O.; Wei, G.; Shi, M.; Tabel, H. CR3 (CD11b/CD18) is the major macrophage receptor for IgM antibody-mediated phagocytosis of African trypanosomes: Diverse effect on subsequent synthesis of tumor necrosis factor alpha and nitric oxide. Microbes Infect. 2006, 8, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Magez, S.; Radwanska, M.; Beschin, A.; Sekikawa, K.; De Baetselier, P. Tumor necrosis factor alpha is a key mediator in the regulation of experimental Trypanosoma brucei infections. Infect. Immun. 1999, 67, 3128–3132. [Google Scholar] [CrossRef] [Green Version]
- Cnops, J.; De Trez, C.; Bulte, D.; Radwanska, M.; Ryffel, B.; Magez, S. IFN-γ mediates early B-cell loss in experimental African trypanosomosis. Parasite Immunol. 2015, 37, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, M.; Guirnalda, P.; De Trez, C.; Ryffel, B.; Black, S.; Magez, S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008, 4, e1000078. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.P.; Wang, H.; Barry, J.D. Mosaic VSGs and the scale of Trypanosoma brucei antigenic variation. PLoS Pathog. 2013, 9, e1003502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gee, A.L.; Levine, R.F.; Mansfield, J.M. Genetics of resistance to the African trypanosomes. VI. Heredity of resistance and variable surface glycoprotein-specific immune responses. J. Immunol. 1988, 140, 283–288. [Google Scholar] [PubMed]
- Dagenais, T.R.; Demick, K.P.; Bangs, J.D.; Forest, K.T.; Paulnock, D.M.; Mansfield, J.M. T-cell responses to the trypanosome variant surface glycoprotein are not limited to hypervariable subregions. Infect. Immun. 2009, 77, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagenais, T.R.; Freeman, B.E.; Demick, K.P.; Paulnock, D.M.; Mansfield, J.M. Processing and presentation of variant surface glycoprotein molecules to T cells in African trypanosomiasis. J. Immunol. 2009, 183, 3344–3355. [Google Scholar] [CrossRef]
- Schleifer, K.W.; Mansfield, J.M. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J. Immunol. 1993, 151, 5492–5503. [Google Scholar]
- Sileghem, M.; Flynn, J.N. Suppression of interleukin 2 secretion and interleukin 2 receptor expression during tsetse-transmitted trypanosomiasis in cattle. Eur. J. Immunol. 1992, 22, 767–773. [Google Scholar] [CrossRef]
- Bailey, J.W.; Smith, D.H. The quantitative buffy coat for the diagnosis of trypanosomes. Trop. Dr. 1994, 24, 54–56. [Google Scholar] [CrossRef]
- Buscher, P.; Mumba Ngoyi, D.; Kabore, J.; Lejon, V.; Robays, J.; Jamonneau, V.; Bebronne, N.; Van der Veken, W.; Bieler, S. Improved Models of Mini Anion Exchange Centrifugation Technique (mAECT) and Modified Single Centrifugation (MSC) for sleeping sickness diagnosis and staging. PLoS Negl. Trop. Dis. 2009, 3, e471. [Google Scholar] [CrossRef] [Green Version]
- Lejon, V.; Buscher, P.; Nzoumbou-Boko, R.; Bossard, G.; Jamonneau, V.; Bucheton, B.; Truc, P.; Lemesre, J.L.; Solano, P.; Vincendeau, P. The separation of trypanosomes from blood by anion exchange chromatography: From Sheila Lanham’s discovery 50 years ago to a gold standard for sleeping sickness diagnosis. PLoS Negl. Trop. Dis. 2019, 13, e0007051. [Google Scholar] [CrossRef]
- Bieler, S.; Matovu, E.; Mitashi, P.; Ssewannyana, E.; Bi Shamamba, S.K.; Bessell, P.R.; Ndung’u, J.M. Improved detection of Trypanosoma brucei by lysis of red blood cells, concentration and LED fluorescence microscopy. Acta Trop. 2012, 121, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Lejon, V.; Büscher, P. Review Article: Cerebrospinal fluid in human African trypanosomiasis: A key to diagnosis, therapeutic decision and post-treatment follow-up. Trop. Med. Int. Health 2005, 10, 395–403. [Google Scholar] [CrossRef]
- Magnus, E.; Vervoort, T.; Van Meirvenne, N. A card-agglutination test with stained trypanosomes (C.A.T.T.) for the serological diagnosis of T. b. gambiense trypanosomiasis. Ann. Soc. Belg. Med. Trop. 1978, 58, 169–176. [Google Scholar] [PubMed]
- Bajyana Songa, E.; Kageruka, P.; Hamers, R. The use of the card agglutination test (Testryp CATT) for the serodiagnosis of T. evansi infection. Ann. Soc. Belg. Med. Trop. 1987, 67, 51–57. [Google Scholar]
- Dama, E.; Camara, O.; Kaba, D.; Koffi, M.; Camara, M.; Compaore, C.; Ilboudo, H.; Courtin, F.; Kabore, J.; N’Gouan, E.K.; et al. Immune trypanolysis test as a promising bioassay to monitor the elimination of gambiense human African trypanosomiasis. Parasite 2019, 26, 68. [Google Scholar] [CrossRef] [Green Version]
- Bisser, S.; Lumbala, C.; Nguertoum, E.; Kande, V.; Flevaud, L.; Vatunga, G.; Boelaert, M.; Buscher, P.; Josenando, T.; Bessell, P.R.; et al. Sensitivity and Specificity of a Prototype Rapid Diagnostic Test for the Detection of Trypanosoma brucei gambiense Infection: A Multi-centric Prospective Study. PLoS Negl. Trop. Dis. 2016, 10, e0004608. [Google Scholar] [CrossRef]
- Lumbala, C.; Matovu, E.; Sendagire, H.; Kazibwe, A.J.N.; Likwela, J.L.; Muhindo Mavoko, H.; Kayembe, S.; Lutumba, P.; Bieler, S.; Van Geertruyden, J.P.; et al. Performance evaluation of a prototype rapid diagnostic test for combined detection of gambiense human African trypanosomiasis and malaria. PLoS Negl. Trop. Dis. 2020, 14, e0008168. [Google Scholar] [CrossRef] [Green Version]
- Büscher, P.; Mertens, P.; Leclipteux, T.; Gilleman, Q.; Jacquet, D.; Mumba-Ngoyi, D.; Pyana, P.P.; Boelaert, M.; Lejon, V. Sensitivity and specificity of HAT Sero-K-SeT, a rapid diagnostic test for serodiagnosis of sleeping sickness caused by Trypanosoma brucei gambiense: A case-control study. Lancet Glob. Health 2014, 2, e359–e363. [Google Scholar] [CrossRef] [Green Version]
- Jamonneau, V.; Camara, O.; Ilboudo, H.; Peylhard, M.; Koffi, M.; Sakande, H.; N’Dri, L.; Sanou, D.; Dama, E.; Camara, M.; et al. Accuracy of individual rapid tests for serodiagnosis of gambiense sleeping sickness in West Africa. PLoS Negl. Trop. Dis. 2015, 9, e0003480. [Google Scholar] [CrossRef] [Green Version]
- Boelaert, M.; Mukendi, D.; Bottieau, E.; Kalo Lilo, J.R.; Verdonck, K.; Minikulu, L.; Barbe, B.; Gillet, P.; Yansouni, C.P.; Chappuis, F.; et al. A Phase III Diagnostic Accuracy Study of a Rapid Diagnostic Test for Diagnosis of Second-Stage Human African Trypanosomiasis in the Democratic Republic of the Congo. EBioMedicine 2018, 27, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Grab, D.J.; Nikolskaia, O.V.; Courtioux, B.; Thekisoe, O.M.M.; Magez, S.; Bogorad, M.; Dumler, J.S.; Bisser, S. Using detergent-enhanced LAMP for African trypanosome detection in human cerebrospinal fluid and implications for disease staging. PLoS Negl. Trop. Dis. 2019, 13, e0007631. [Google Scholar] [CrossRef]
- Nikolskaia, O.V.; Thekisoe, O.M.; Dumler, J.S.; Grab, D.J. Loop-Mediated Isothermal Amplification for Detection of the 5.8S Ribosomal Ribonucleic Acid Internal Transcribed Spacer 2 Gene Found in Trypanosoma brucei gambiense. Am. J. Trop. Med. Hyg. 2017, 96, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Kuboki, N.; Inoue, N.; Sakurai, T.; Di Cello, F.; Grab, D.J.; Suzuki, H.; Sugimoto, C.; Igarashi, I. Loop-mediated isothermal amplification for detection of African trypanosomes. J. Clin. Microbiol. 2003, 41, 5517–5524. [Google Scholar] [CrossRef] [Green Version]
- Berthier, D.; Breniere, S.F.; Bras-Goncalves, R.; Lemesre, J.L.; Jamonneau, V.; Solano, P.; Lejon, V.; Thevenon, S.; Bucheton, B. Tolerance to Trypanosomatids: A Threat, or a Key for Disease Elimination? Trends Parasitol. 2016, 32, 157–168. [Google Scholar] [CrossRef]
- Tong, Q.; Chen, R.; Kong, Q.; Goossens, J.; Radwanska, M.; Lou, D.; Ding, J.; Zheng, B.; Fu, Y.; Wang, T.; et al. DNA detection of Trypanosoma evansi: Diagnostic validity of a new assay based on loop-mediated isothermal amplification (LAMP). Vet. Parasitol. 2018, 250, 1–6. [Google Scholar] [CrossRef]
- Njiru, Z.K.; Constantine, C.C.; Masiga, D.K.; Reid, S.A.; Thompson, R.C.; Gibson, W.C. Characterization of Trypanosoma evansi type B. Infect. Genet. Evol. 2006, 6, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Njiru, Z.K.; Ouma, J.O.; Enyaru, J.C.; Dargantes, A.P. Loop-mediated Isothermal Amplification (LAMP) test for detection of Trypanosoma evansi strain B. Exp. Parasitol. 2010, 125, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Pinto Torres, J.E.; Goossens, J.; Stijlemans, B.; Sterckx, Y.G.; Magez, S. Development of a recombinase polymerase amplification lateral flow assay for the detection of active Trypanosoma evansi infections. PLoS Negl. Trop. Dis. 2020, 14, e0008044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto Torres, J.E.; Goossens, J.; Ding, J.; Li, Z.; Lu, S.; Vertommen, D.; Naniima, P.; Chen, R.; Muyldermans, S.; Sterckx, Y.G.; et al. Development of a Nanobody-based lateral flow assay to detect active Trypanosoma congolense infections. Sci. Rep. 2018, 8, 9019. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.; Odongo, S.; Lee, F.; Gaspariunaite, V.; Muyldermans, S.; Magez, S.; Sterckx, Y.G. Structural basis for the high specificity of a Trypanosoma congolense immunoassay targeting glycosomal aldolase. PLoS Negl. Trop. Dis. 2017, 11, e0005932. [Google Scholar] [CrossRef] [Green Version]
- Odongo, S.; Sterckx, Y.G.; Stijlemans, B.; Pillay, D.; Baltz, T.; Muyldermans, S.; Magez, S. An Anti-proteome Nanobody Library Approach Yields a Specific Immunoassay for Trypanosoma congolense Diagnosis Targeting Glycosomal Aldolase. PLoS Negl. Trop. Dis. 2016, 10, e0004420. [Google Scholar] [CrossRef] [PubMed]
- Obishakin, E.; Stijlemans, B.; Santi-Rocca, J.; Vandenberghe, I.; Devreese, B.; Muldermans, S.; Bastin, P.; Magez, S. Generation of a nanobody targeting the paraflagellar rod protein of trypanosomes. PLoS ONE 2014, 9, e115893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Pinto Torres, J.E.; Goossens, J.; Vertommen, D.; Caljon, G.; Sterckx, Y.G.; Magez, S. An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi. Vaccines (Basel) 2020, 8, 415. [Google Scholar] [CrossRef]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. The Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Yun, O.; Priotto, G.; Tong, J.; Flevaud, L.; Chappuis, F. NECT is next: Implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neau, P.; Hanel, H.; Lameyre, V.; Strub-Wourgaft, N.; Kuykens, L. Innovative Partnerships for the Elimination of Human African Trypanosomiasis and the Development of Fexinidazole. Trop. Med. Infect. Dis. 2020, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Dickie, E.A.; Giordani, F.; Gould, M.K.; Maser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Infect. Dis. 2020, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; HP, D.E.K.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef]
- Peregrine, A.S.; Mamman, M. Pharmacology of diminazene: A review. Acta Trop. 1993, 54, 185–203. [Google Scholar] [CrossRef]
- Black, S.J.; Mansfield, J.M. Prospects for vaccination against pathogenic African trypanosomes. Parasite Immunol. 2016, 38, 735–743. [Google Scholar] [CrossRef]
- Lejon, V.; Mumba Ngoyi, D.; Kestens, L.; Boel, L.; Barbe, B.; Kande Betu, V.; van Griensven, J.; Bottieau, E.; Muyembe Tamfum, J.J.; Jacobs, J.; et al. Gambiense human african trypanosomiasis and immunological memory: Effect on phenotypic lymphocyte profiles and humoral immunity. PLoS Pathog. 2014, 10, e1003947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejon, V.; Ngoyi, D.M.; Ilunga, M.; Beelaert, G.; Maes, I.; Buscher, P.; Fransen, K. Low specificities of HIV diagnostic tests caused by Trypanosoma brucei gambiense sleeping sickness. J. Clin. Microbiol. 2010, 48, 2836–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, L.D.; Juyal, P.D.; Sharma, N.S. Immune responses to haemorrhagic septicaemia (HS) vaccination in Trypanosoma evansi infected buffalo-calves. Trop. Anim. Health Prod. 2010, 42, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.G.; Do, T.T.; Huong, N.T.; Dung, N.T.; Thanh, N.G.; Vercruysse, J.; Goddeeris, B.M. The effect of Trypanosoma evansi infection on pig performance and vaccination against classical swine fever. Vet. Parasitol. 2003, 111, 115–123. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magez, S.; Pinto Torres, J.E.; Oh, S.; Radwanska, M. Salivarian Trypanosomes Have Adopted Intricate Host-Pathogen Interaction Mechanisms That Ensure Survival in Plain Sight of the Adaptive Immune System. Pathogens 2021, 10, 679. https://doi.org/10.3390/pathogens10060679
Magez S, Pinto Torres JE, Oh S, Radwanska M. Salivarian Trypanosomes Have Adopted Intricate Host-Pathogen Interaction Mechanisms That Ensure Survival in Plain Sight of the Adaptive Immune System. Pathogens. 2021; 10(6):679. https://doi.org/10.3390/pathogens10060679
Chicago/Turabian StyleMagez, Stefan, Joar Esteban Pinto Torres, Seoyeon Oh, and Magdalena Radwanska. 2021. "Salivarian Trypanosomes Have Adopted Intricate Host-Pathogen Interaction Mechanisms That Ensure Survival in Plain Sight of the Adaptive Immune System" Pathogens 10, no. 6: 679. https://doi.org/10.3390/pathogens10060679